

 下载:
下载:

-
Type 2 diabetes (T2DM) is a common metabolic disorder associated with environmental and genetic factors characterized by both insulin resistance and relative insufficiency of insulin secretion. T2DM has a high genetic predisposition. It is reported that 40% of first-degree relatives of patients with T2DM may develop diabetes; however, the incidence rate is only 6% in the general population[1]. Hyperlipidemia is occasionally accompanied by diabetes, and although patients with type 2 diabetes and hyperlipidemia are not rare, cases of type 2 diabetes and hyperlipidemia caused by gene mutation are rarely reported. The increasing use of gene sequencing increases the accuracy of clinical diagnosis in clinical practice.
The perilipin gene (PLIN1, also known as perinatal lipoprotein 1) is primarily expressed in adipocytes, playing an important role in lipid droplet formation and regulation of triglyceride metabolism[2]. The mutation of this gene is associated with familial partial lipodystrophy type 4 (FPLD4)[3], severe insulin resistance, type 2 diabetes, extreme hypertriglyceridemia, and nonalcoholic fatty liver disease[4]. Additionally, Serine/Threonine Protein Kinase 2 (AKT2) mediates the physiological effects of insulin[5]. It has been previously reported that the mutation of AKT2 is correlated with various disorders of insulin action, including severe insulin resistance, hypoinsulinemia, hypoketosis, and hypolipemia[5-7]. In this study, we presented a unique case of two distinct heterozygous mutations in the AKT2 and PLIN1 genes that together caused insulin resistance, diabetes, and dyslipidemia in the patient.
An unreported missense mutation of heterozygous PLIN1 and AKT2 genes was observed in the patient's genetic testing results. To date, insulin resistance, diabetes, and hyperlipidemia caused by the AKT2 (c.1202A>G) gene mutation combined with the PLIN1 (c.1565G>A) gene mutation have not been reported. This is the first case of diabetes and hyperlipidemia caused by the AKT2 gene mutation combined with the PLIN1 gene mutation.
A 24-year-old Chinese female patient with hyperglycemia and increased blood ketone was admitted to the Department of Endocrinology and Metabolism of the Second Affiliated Hospital of Chongqing Medical University. She had nausea and vomited once daily prior to admission. She had been diagnosed with polydipsia (> 3 L/d), polyuria with high blood glucose (over 33.3 mmol/L by capsular blood sugar), and ketonuria at 14 years of age at a local hospital. She had been treated with insulin and metformin for years. She had experienced three episodes of diabetic ketoacidosis prior to admission. She had a history of hypertriglyceridemia (triglyceride, 10−30 mmol/L) and acute pancreatitis at 19 years of age. Menarche occurred at 12 years of age and had been regular since then. She had no history of high blood pressure. Her father had diabetes.
On admission, her body mass index (weight, calculated in kg, divided by the square of her height, calculated in m) was 22.8 kg/m2. No significant muscle appearance, lipopenia, acanthosis nigricans, and hirsutism were observed in the clinical examination.
Laboratory evaluation revealed that fasting blood glucose was 16.26 mmol/L, glycosylated hemoglobin A1c increased by 11.8%, serum β-hydroxybutyric acid increased by 2.27 mmol/L (reference range, 0.02−0.27 mmol/L), and arterial pH 7.34. Total cholesterol increased by 10.12 mmol/L (reference range, 0.00−5.18 mmol/L); triglyceride level was 32.35 mmol/L (reference range, 0.00−1.70 mmol/L); low-density lipoprotein was 5.29 mmol/L (reference range, 0.00−3.37 mmol/L); the free fatty acid level was 1.06 mmol/L (reference range, 0.10−0.45 mmol/L); apolipoprotein A1 level was 0.64 g/L (reference range, 1.00−1.60 g/L); apolipoprotein B level was 1.82 g/L (reference range, 0.60−1.10 g/L); apolipoprotein E level was 86.1 mg/L (reference range, 27.0−49.0 mg/L); the patient’s pancreatic enzyme levels were normal. The glutamic acid decarboxylase antibody and insulin autoantibody were normal. Her albumin was 53.3 g/L in the normal range, creatinine level was 60.8 µmol/L in the normal range, aspartate aminotransferase was 36 U/L (reference range, 13−35 U/L) higher, urinary microalbumin was 27.6 mg/L (reference range, 0−15 mg/L), and urinary albumin/creatinine ratio was 6.33 mg/mmol (reference range, 0−3 mg/mmol). Diabetic complication screening confirmed diabetic retinopathy, diabetic peripheral neuropathy, and diabetic nephropathy after being cured of diabetic ketoacidosis. Her visceral fat area was 57 cm2, and her subcutaneous fat area was 108 cm2. The abdominal color ultrasound depicted fatty liver, and the plasma cortisol and serum adrenocorticotropic hormone levels were normal. Carotid artery ultrasound exhibited no obvious abnormality, no arterial plaque, and vessel stenosis.
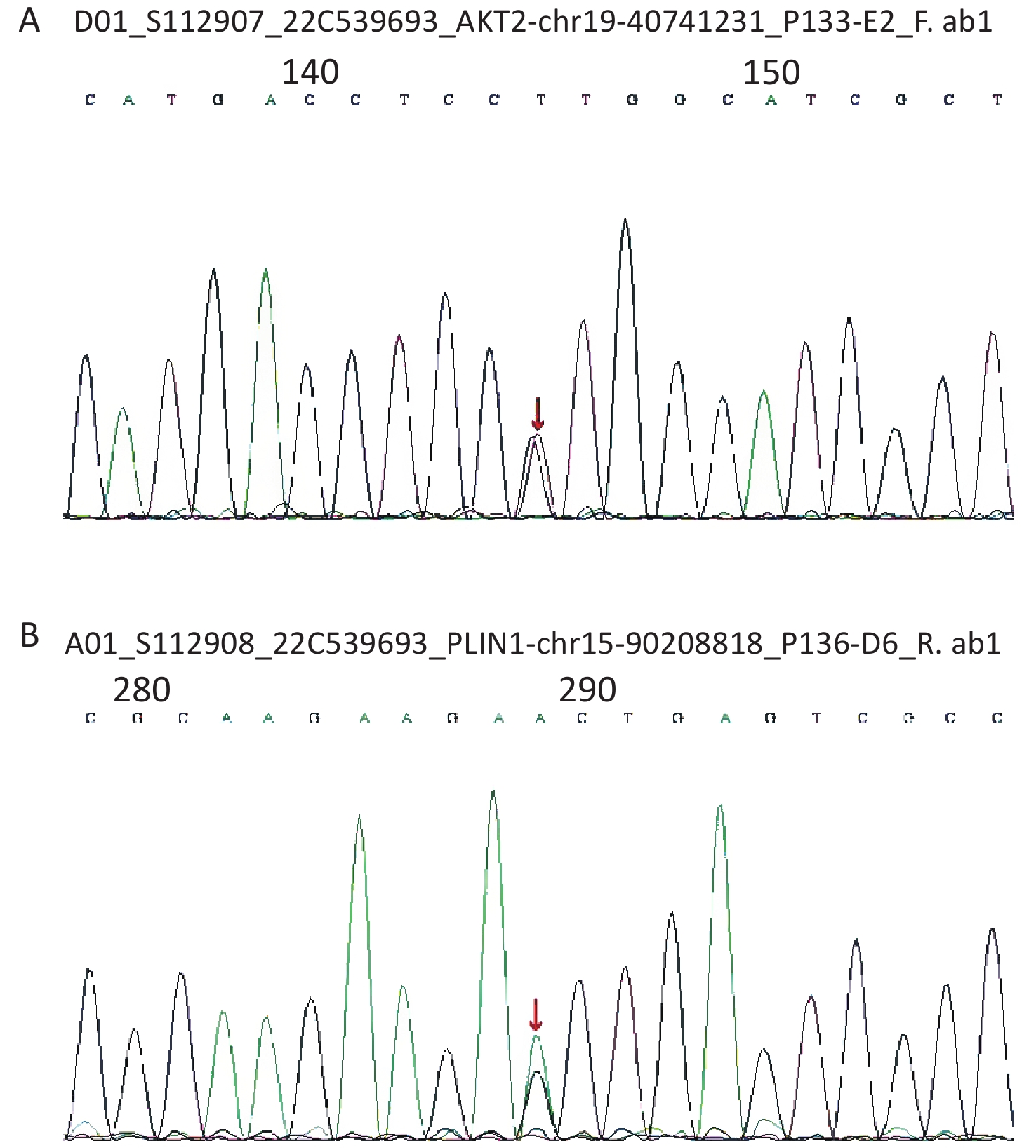
Considering the early onset of metabolic abnormalities, whole exome sequencing (WES015 GenCap® V6.0) was performed to detect mutations in the blood sample of the patient. She had a heterozygous mutation on exon 9 (c.1565G>A) of the PLIN1 gene (Figure 1), which predicted an incorrect translation causing the synthesis of an abnormal amino acid at position 522 (p. Ser522Asn). She also had a heterozygous mutation on exon 12 (c.1202A>G) of the AKT2 gene (Figure 1), which predicted an incorrect translation causing the synthesis of 401 anomalous amino acids (p. Lys401Arg).
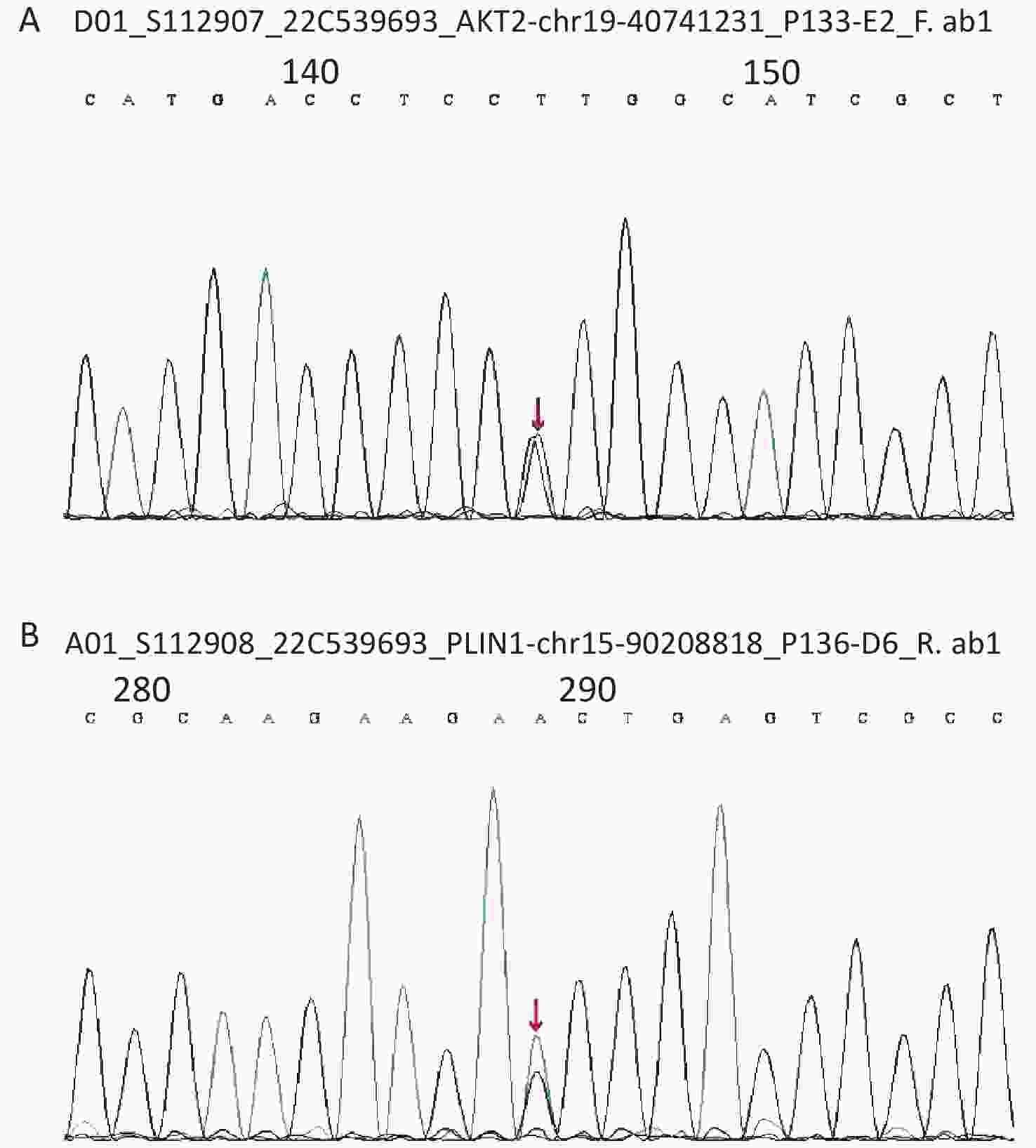
Figure 1. (A) The sequence of AKT2 gene. (B) The sequence of PLIN1 gene.
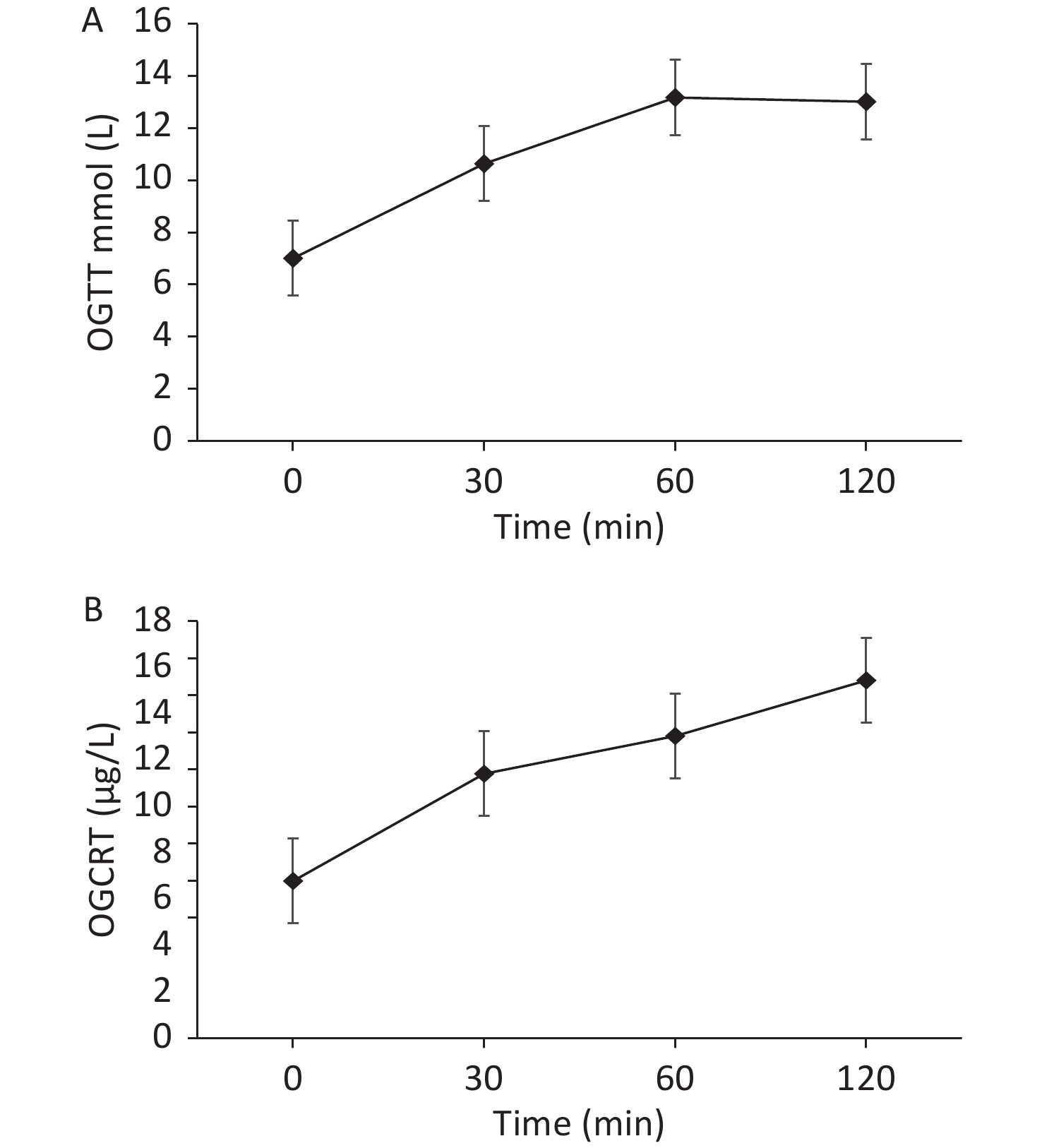
On admission, we provided a saline replacement, regular insulin to lower the blood glucose, and glucose together with insulin to the patient to inhibit lipolysis. Further, on day 2 of admission, her serum β-hydroxybutyric acid returned to normal. Afterward, we administered continuous subcutaneous insulin infusion to the patient for further 3 days. An oral glucose tolerance test was performed to evaluate the beta cell function when the fasting blood glucose was < 8 mmol/L. The results are presented in Figure 2.
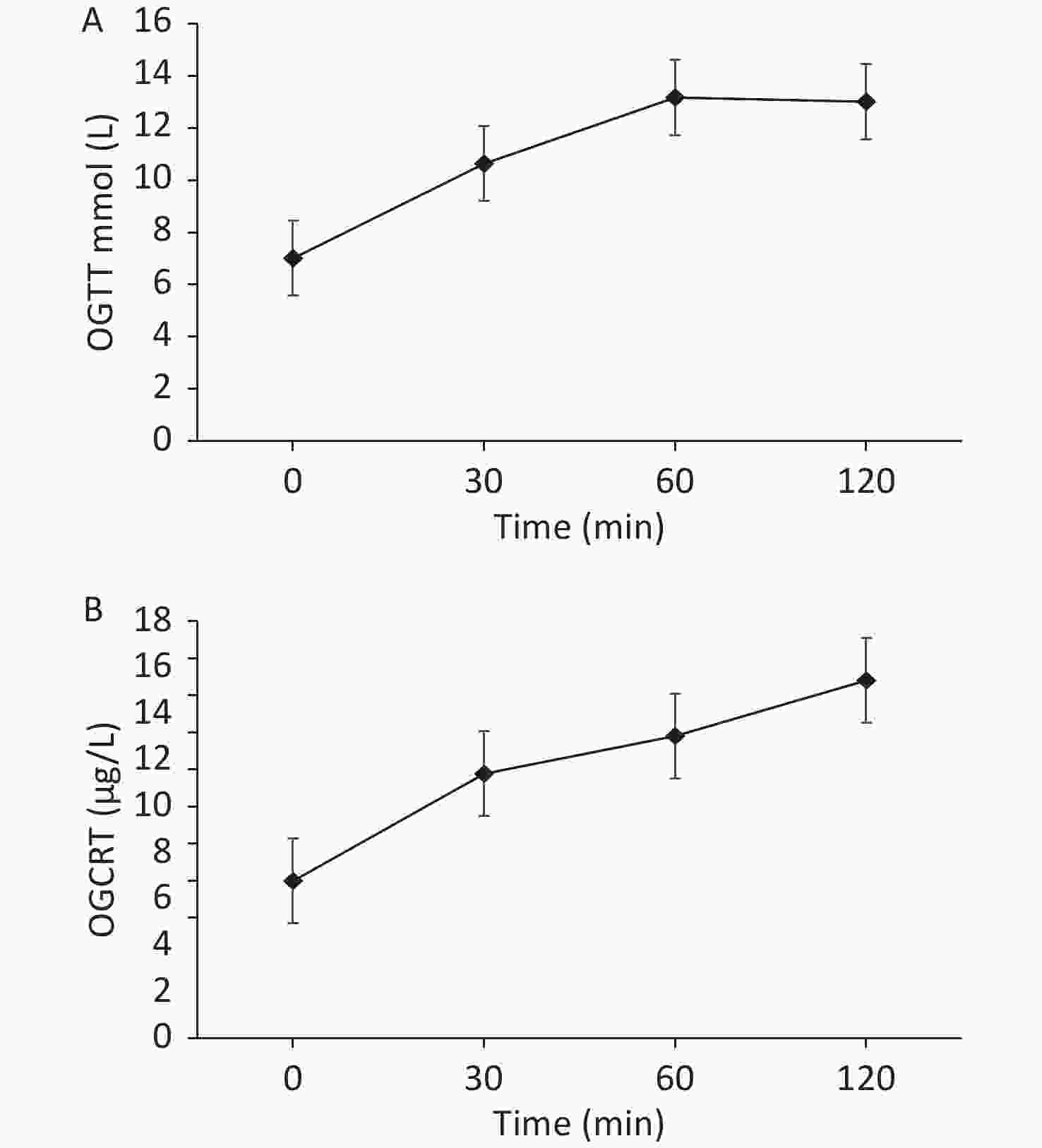
Figure 2. (A) The patient’s oral glucose tolerance test results of 75 g glucose. (B) The patient’s C-peptide reaction test result.
Based on the beta cell function of the patient, the continuous subcutaneous insulin infusion was changed to insulin detemir before bedtime, combined with 30 mg pioglitazone once daily, 1.5 g metformin once a day, 100 mg canagliflozin once a day, 50 mg acarbose thrice a day to control blood glucose, and 200 mg fenofibrate once a day to treat hypertriglyceridemia. During dismission, her fasting blood glucose fluctuated at 6−7 mmol/L, plasma triglyceride decreased but still exceeded the normal level (i.e. 8.79 mmol/L), and total cholesterol decreased to the normal level i.e. 4.75 mmol/L.
We demonstrated a rare case of diabetes at a young age, characterized by repeated ketosis, insulin resistance, and severe hyperlipidemia. Further genetic sequencing revealed that the patient had a heterozygous missense mutation in both the AKT2 (c.1202A>G) gene and PLIN1 (c.1565G>A) gene. The pathogenesis of this mutation remains elusive; however, considering the patient's morbidity characteristics, family history of diabetes, negative glutamic acid decarboxylase antibody, and insulin autoantibody, we conclude that PLIN1 and AKT2 gene mutations caused insulin resistance, diabetes, and severe hyperlipidemia.
PLIN1 is the most abundant phosphoprotein in adipocytes that binds confirmative to the phospholipid surface monolith of lipid droplets and regulates lipases, such as adipose tissue triglyceride lipase and hormone-sensitive lipase[4]. It has bidirectional regulation of lipolysis and plays an important role in the dynamic regulation of lipolysis. PLIN1 phosphorylation was low under basic conditions, which reduced TG hydrolysis. In the low phosphorylation state, PLIN1 acts as a valve, preventing lipase from entering the neutral lipid droplet. In contrast, PLIN1 actively promotes lipase activity following phosphorylation[8]. The PLIN1 mutation was first identified in patients with FPLD4 (familial partial lipodystrophy type 4), and the main characteristics of those patients included a lack of peripheral fat pool, severe insulin resistance, and extreme hypertriglyceridemia[4]. Mutations in the PLIN1 gene (P. Val398fs) or (p. Leu404fs) are also associated with severe insulin resistance and extreme hypertriglyceridemia[9]. Renal involvement in the patient with first type 4 FPLD is related to the (p. Y401Lfs) mutation of the PLIN1 gene[10]. The mutation of the PLIN1 (c.1565G>A) gene has not been reported. Combined with the characteristics of patients, we speculated that PLIN1 (c.1565G>A) may be pathogenic.
AKT2 is a serine/threonine protein kinase that is involved in the downstream of insulin receptors and mediates the physiological effects of insulin. AKT2 plays key roles not only in insulin action but also in blood lipid metabolism and regulation. Post-receptor insulin resistance caused by AKT2 mutations not only increases the fasting blood glucose but also increases the very low-density lipid cholesterol/total cholesterol ratio, decreases high-density lipid cholesterol level, and increases low-density lipid cholesterol[6]. The AKT2 gene mutation has been reported in familial insulin resistance and diabetes. It has been reported that the R274H mutation of AKT2 causes severe insulin resistance, significant hyperinsulinemia, and diabetes[6] whereas the Glu17Lys mutations of AKT2 cause hypoglycemia, and c.49G3A (p.E17K) mutations of AKT2 cause hypoinsulinemia, hypoketosis, and hypolipidaemia[5, 7] (Table 1). In this case study, the phenotypes were severe insulin resistance, diabetes, and hyperlipidemia. Meanwhile, the genetic test revealed mutations in two genes, AKT2 (c.1202A>G) and PLIN1 (c.1565G>A). We speculated that it may be owing to the combined effect of these two genes; however, considering the greater influence of the PLIN1 gene on insulin resistance and blood lipid metabolism and regulation, it may also be caused by the heterozygous mutation of PLIN1. Unfortunately, the patient’s parents do not stay with her, and we could not obtain additional information from them.
Table 1. Characteristics of individuals with PLIN1 or AKT2 mutations
Reference ARYA V. B.[5], 2014 Kristina Kozusko[4], 2015 HUSSAIN K[7], 2011 JERU I[3], 2019 PLNI1 − c.1298_1299delTG (p439fs) − − − c.1191_1192del
(p.Val398Glyfs*166)AKT2 c.49G3A
(p.E17K)− − c.49G>A
(p.Glu17Lys)c.49G>A
(p.Glu17Lys)c.49G>A (p.Glu17Lys) − Sex Female Female Male Male Male Female Female Age of onset 8 months 41 years 15 years 1 year 15 months 5 months 20 years Race Caucasian Caucasian Caucasian European European European European French BMI (kg/m2) − 31.2 25.1 − − − 23 Insulin (μU/mL) 2 ND 40.92 < 1 < 0.4 < 0.3 NA Glucose (mmol/L) 2.4 11.3 5.1 2.2 1.6 1.6 8 HbA1c (%) − 7.9 5.1 − − − 8 TG (mmol/L) − 56.1 26.3 − − − 2 TC (mmol/L) − 10.5 4.4 − − − − Family history − diabetes − − − hypoglycemia − Characteristics Hemihypertrophy and symptomatic hypoglycemia Type 2 diabetes, extreme hypertriglyceridemia, recurrent pancreatitis, nonalcoholic fatty liver disease (NAFLD), limb and femoro gluteal lipodystrophy Acanthosis nigricans, hepatomegaly, NAFLD, a striking paucity of limb fat, severe hypertriglyceridemia, extreme insulin resistance Left sided hemihypertrophy, grand mal Seizures, severe spontaneous hypoglycemia Hypotonic, with poor tolerance of fasting and episodes of pallor and lethargy before food, and was still waking at night for feeding Hypoglycemia, Mild facial asymmetry Android habitus; Four limb lipoatrophy, muscular hypertrophy, liver steatosis, hirsutism; Oligomenorrhea, fatigue; Unexplained recurrent vomiting, permanent diabetes Reference JERU I.[3], 2019 Sheetal Gandotra, Ph.D.[9], 2011 CHEN R. X.[10], 2018 SEMPLE R. K.[6], 2009 2022 PLNI1 c.1202_1205dup (p.Pro403Argfs*164) c.1210-1G→T
(p.Leu404fs)c.1191_1192delAG
(p.Val398fs)c.1191_1192delAG
(p.Val398fs)c.1201_1202insT
(p.Y401Lfs*165)− c.1565G>A
(p.Ser522Asn)AKT2 − − − − − c.821G>A (p.Arg274His) c.1202A>G
(p.Lys401Arg)Sex Female Female Female Female Female Female Female Age of onset 30 years 23 years 25 years 21 years 12 years 40 years 14 years Race French French French French Chinese Caucasian Chinese BMI (kg/m2) 30.7 23 24.7 26.9 22.9 26.7 22.8 Reference JERU I.[3], 2019 Sheetal Gandotra, Ph.D.[9], 2011 CHEN R. X.[10], 2018 SEMPLE R. K.[6], 2009 2022 Insulin (µU/ML) 16.08 29.9 − 27.997 172.4 15.08 − Glucose (mmol/L) 7.4 6.1 11.11 16 5.8 15.7 16.26 HbA1c (%) 6.5 − 7.9 − 5.6 − 11.8 TG (mmol/L) 5.7 29.4 9.0 30 3.4 4.8 32.35 TC (mmol/L) − − − − − 5.4 10.12 Family history − − − − insulin-resistant diabetes diabetes diabetes Characteristics Lipoatrophy, muscular hypertrophy, Acanthosis nigricans, Severe fatigue; Hypertension, permanent diabetes Severe hypertriglyceridemia, insulin resistance, hypertension, diabetes and liver steatosis, lipoatrophy, muscular hypertrophy, acanthosis nigricans Severe hypertension, acanthosis nigricans, hypertriglyceridemia, insulin resistance, diabetes, acromegaloid facial appearance , muscular hypertrophy Insulin-resistant diabetes with extensive acanthosis nigricans, severe hypertriglyceridemia, steatohepatitis, cushingoid appearance Proteinuria, hypertriglyceridemia, nonalcoholic steatohepatitis, insulin-resistant diabetes, polycystic ovaries, acanthosis nigricans Severe insulin resistance, hyperglycemia, lipodystrophy High blood glucose, blood ketone, severe insulin resistance, and hyperlipidemia, nausea, vomiting Type 2 diabetes combined with hyperlipidemia is a common metabolic disorder; however, there have been few reported cases of genetic factors involved in this disorder. In this study, we reported the first case of a young female patient with diabetes and hyperlipidemia caused by heterozygous missense mutations of both PLIN1 and AKT2. PLIN1, AKT2, and other related gene tests should be performed in young patients with diabetes complicated with severe hyperlipidemia and genetic predisposition to further clarify the pathogenicity and guide future treatment.
LIU Jing contributed to the drafting of the manuscript. LI Sheng Bing, LUO Xie, YUAN Lei, and LAI Ye Rui contributed to the analysis of the data and approved the final version of the manuscript. ZHANG Li Li contributed to the drafting of the manuscript and critical discussion and approved the final version of the manuscript.
We thank all the participants and the patient in the study.
The authors declare that they have no competing interests.
doi: 10.3967/bes2023.043
Type 2 Diabetes and Hyperlipidemia Caused by AKT2 Gene Combined with PLIN1 Gene Mutation: A Case Report
-
-
Figure 2. (A) The patient’s oral glucose tolerance test results of 75 g glucose. (B) The patient’s C-peptide reaction test result.
Table 1. Characteristics of individuals with PLIN1 or AKT2 mutations
Reference ARYA V. B.[5], 2014 Kristina Kozusko[4], 2015 HUSSAIN K[7], 2011 JERU I[3], 2019 PLNI1 − c.1298_1299delTG (p439fs) − − − c.1191_1192del
(p.Val398Glyfs*166)AKT2 c.49G3A
(p.E17K)− − c.49G>A
(p.Glu17Lys)c.49G>A
(p.Glu17Lys)c.49G>A (p.Glu17Lys) − Sex Female Female Male Male Male Female Female Age of onset 8 months 41 years 15 years 1 year 15 months 5 months 20 years Race Caucasian Caucasian Caucasian European European European European French BMI (kg/m2) − 31.2 25.1 − − − 23 Insulin (μU/mL) 2 ND 40.92 < 1 < 0.4 < 0.3 NA Glucose (mmol/L) 2.4 11.3 5.1 2.2 1.6 1.6 8 HbA1c (%) − 7.9 5.1 − − − 8 TG (mmol/L) − 56.1 26.3 − − − 2 TC (mmol/L) − 10.5 4.4 − − − − Family history − diabetes − − − hypoglycemia − Characteristics Hemihypertrophy and symptomatic hypoglycemia Type 2 diabetes, extreme hypertriglyceridemia, recurrent pancreatitis, nonalcoholic fatty liver disease (NAFLD), limb and femoro gluteal lipodystrophy Acanthosis nigricans, hepatomegaly, NAFLD, a striking paucity of limb fat, severe hypertriglyceridemia, extreme insulin resistance Left sided hemihypertrophy, grand mal Seizures, severe spontaneous hypoglycemia Hypotonic, with poor tolerance of fasting and episodes of pallor and lethargy before food, and was still waking at night for feeding Hypoglycemia, Mild facial asymmetry Android habitus; Four limb lipoatrophy, muscular hypertrophy, liver steatosis, hirsutism; Oligomenorrhea, fatigue; Unexplained recurrent vomiting, permanent diabetes Reference JERU I.[3], 2019 Sheetal Gandotra, Ph.D.[9], 2011 CHEN R. X.[10], 2018 SEMPLE R. K.[6], 2009 2022 PLNI1 c.1202_1205dup (p.Pro403Argfs*164) c.1210-1G→T
(p.Leu404fs)c.1191_1192delAG
(p.Val398fs)c.1191_1192delAG
(p.Val398fs)c.1201_1202insT
(p.Y401Lfs*165)− c.1565G>A
(p.Ser522Asn)AKT2 − − − − − c.821G>A (p.Arg274His) c.1202A>G
(p.Lys401Arg)Sex Female Female Female Female Female Female Female Age of onset 30 years 23 years 25 years 21 years 12 years 40 years 14 years Race French French French French Chinese Caucasian Chinese BMI (kg/m2) 30.7 23 24.7 26.9 22.9 26.7 22.8 Reference JERU I.[3], 2019 Sheetal Gandotra, Ph.D.[9], 2011 CHEN R. X.[10], 2018 SEMPLE R. K.[6], 2009 2022 Insulin (µU/ML) 16.08 29.9 − 27.997 172.4 15.08 − Glucose (mmol/L) 7.4 6.1 11.11 16 5.8 15.7 16.26 HbA1c (%) 6.5 − 7.9 − 5.6 − 11.8 TG (mmol/L) 5.7 29.4 9.0 30 3.4 4.8 32.35 TC (mmol/L) − − − − − 5.4 10.12 Family history − − − − insulin-resistant diabetes diabetes diabetes Characteristics Lipoatrophy, muscular hypertrophy, Acanthosis nigricans, Severe fatigue; Hypertension, permanent diabetes Severe hypertriglyceridemia, insulin resistance, hypertension, diabetes and liver steatosis, lipoatrophy, muscular hypertrophy, acanthosis nigricans Severe hypertension, acanthosis nigricans, hypertriglyceridemia, insulin resistance, diabetes, acromegaloid facial appearance , muscular hypertrophy Insulin-resistant diabetes with extensive acanthosis nigricans, severe hypertriglyceridemia, steatohepatitis, cushingoid appearance Proteinuria, hypertriglyceridemia, nonalcoholic steatohepatitis, insulin-resistant diabetes, polycystic ovaries, acanthosis nigricans Severe insulin resistance, hyperglycemia, lipodystrophy High blood glucose, blood ketone, severe insulin resistance, and hyperlipidemia, nausea, vomiting  下载: 导出CSV
下载: 导出CSV
-
[1] Wu YL, Ding YP, Tanaka Y, et al. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int J Med Sci, 2014; 11, 1185−200. doi: 10.7150/ijms.10001 [2] Grahn THM, Zhang Y, Lee MJ, et al. FSP27 and PLIN1 interaction promotes the formation of large lipid droplets in human adipocytes. Biochem Biophys Res Commun, 2013; 432, 296−301. doi: 10.1016/j.bbrc.2013.01.113 [3] Jéru I, Vantyghem MC, Bismuth E, et al. Diagnostic challenge in PLIN1-associated familial partial lipodystrophy. J Clin Endocrinol Metab, 2019; 104, 6025−32. doi: 10.1210/jc.2019-00849 [4] Kozusko K, Tsang VHM, Bottomley W, et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes, 2015; 64, 299−310. doi: 10.2337/db14-0104 [5] Arya VB, Flanagan SE, Schober E, et al. Activating AKT2 mutation: hypoinsulinemic hypoketotic hypoglycemia. J Clin Endocrinol Metab, 2014; 99, 391−4. doi: 10.1210/jc.2013-3228 [6] Semple RK, Sleigh A, Murgatroyd PR, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest, 2009; 119, 315−22. [7] Hussain K, Challis B, Rocha N, et al. An activating mutation of AKT2 and human hypoglycemia. Science, 2011; 334, 474. doi: 10.1126/science.1210878 [8] Li SJ, Khan R, Raza SHA, et al. Function and characterization of the promoter region of perilipin 1 (PLIN1): roles of E2F1, PLAG1, C/EBPβ, and SMAD3 in bovine adipocytes. Genomics, 2020; 112, 2400−9. doi: 10.1016/j.ygeno.2020.01.012 [9] Gandotra S, Le Dour C, Bottomley W, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med, 2011; 364, 740−8. doi: 10.1056/NEJMoa1007487 [10] Chen RX, Zhang L, Ye W, et al. The renal manifestations of type 4 familial partial lipodystrophy: a case report and review of literature. BMC Nephrol, 2018; 19, 111. doi: 10.1186/s12882-018-0913-6 -

 点击查看大图
点击查看大图
图(2) / 表ll (1)
计量
- 文章访问数: 903
- HTML全文浏览量: 436
- PDF下载量: 71
- 被引次数: 0

 Quick Links
Quick Links