

 下载:
下载:

-
Global air pollution has become a major threat to human health. Daily variations in air pollution have been linked to increased risks of cardiopulmonary-related morbidity and mortality. In particular, fine particulate matter (PM) with an aerodynamic diameter ≤ 2.5 μm (PM2.5), which mostly originates from coal combustion, vehicle exhaust, and open sources in Chinese cities, is detrimental to health[1]. Epidemiological and experimental investigations have demonstrated that long-term exposure to PM2.5, the most dangerous particulate in the Earth’s atmosphere, is a pivotal risk factor for cardiorespiratory diseases.
Long-term exposure to PM2.5 induces an excess of reactive oxygen species (ROS) and proinflammatory cytokine production, which are critical for PM2.5-mediated toxicity. Studies on human subjects and animal models have shown that PM2.5 exposure results in local and systemic inflammation[2]. However, the molecular mechanisms underlying the PM2.5-induced inflammatory response have not been elucidated. Nuclear factor NF-(κB) signaling, which is crucial for inflammation, is activated by numerous factors, such as toll-like receptor (TLR) ligands of lipopolysaccharides (LPS), hypoxia, and tumor necrosis factor (TNF)-α. Furthermore, excessive ROS production can activate the inhibitor of NF-κB (IκB) kinase (IKK) isoforms, including IKKα and IKKβ. These isoforms phosphorylate IκBα to release transcription factors such as NF-kBp65 and subsequently promote its nuclear translocation, leading to the production of proinflammatory cytokines such as interleukin (IL)-6 and IL-8[3]. Moreover, the TLR protein family plays a fundamental role in pathogen recognition and innate immune activation. Various TLRs exhibit different patterns of expression, and upon activation by stimuli, they mediate immune responses by triggering a cascade of distinct events. TLR4, the receptor that mobilizes NF-κB, and promotes inflammatory cytokine production, has been proven to play a key role in inflammatory responses, which directly activates downstream signaling pathways by interacting with myeloid differentiation primary response 88 (MyD88), further degrading IκBα and decreasing phosphorylated NF-κB levels [4]. Therefore, in the present study, we investigated the toxicological properties of PM2.5 collected in an urban area of China (Changchun) and elucidated the underlying toxicological mechanisms, such as the regulation of the expression of NF-κB, MyD88, and TLR4 proteins, focusing mainly on inflammation and oxidative stress in macrophages. Our findings may provide new insights into the molecular mechanisms of PM2.5 exposure toxicity and aid in the design of new therapeutic strategies to prevent PM2.5-induced toxicity.
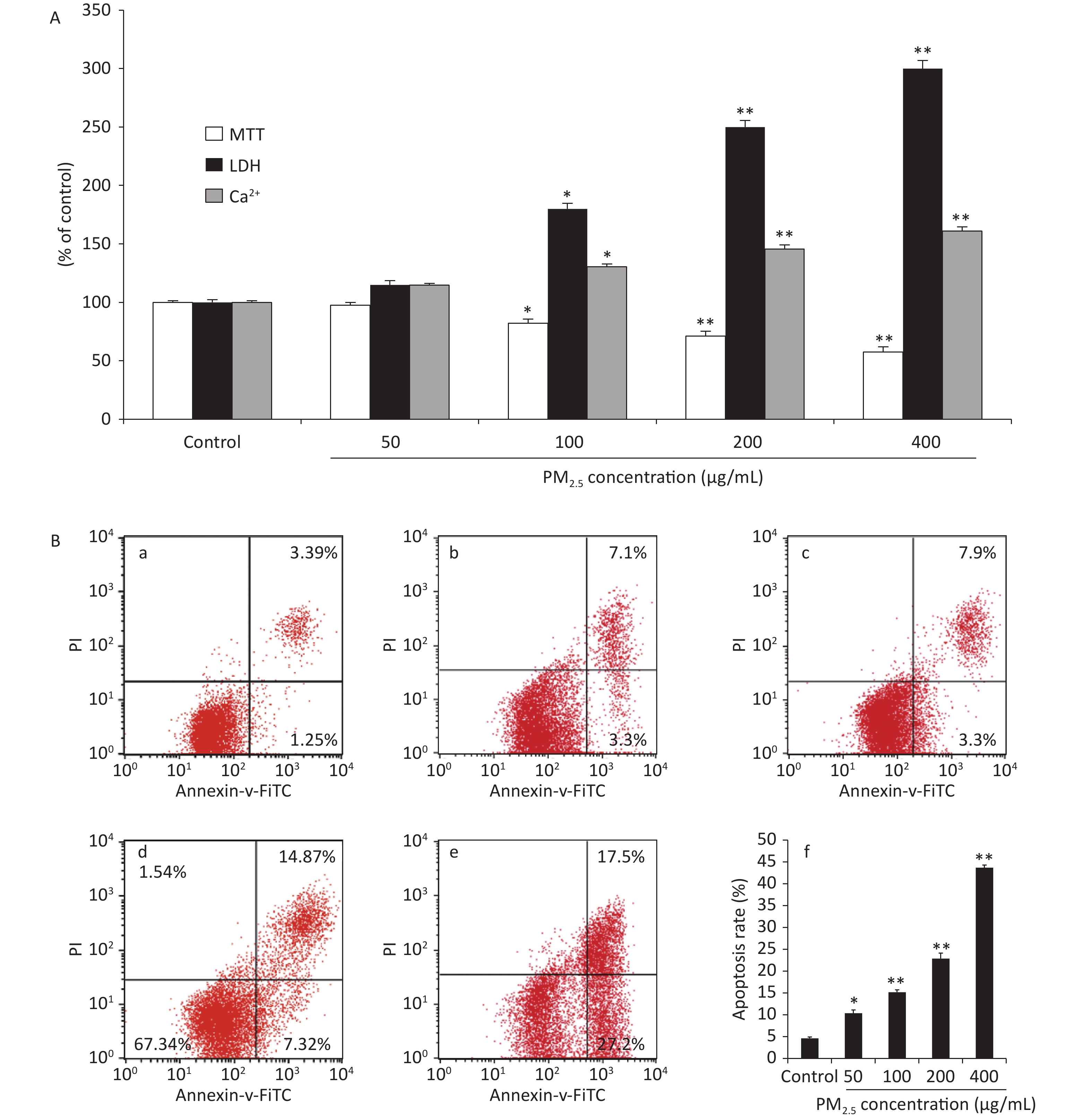
The cell viability measured using the methyl thiazolyl tetrazolium (MTT) reduction assays showed concentration-dependent decreases following PM2.5 exposures in RAW264.7 cells, which could be attributed to an increase in extracellular lactate dehydrogenase (LDH) release from the macrophages. The MTT reduction assay measures the redox activity of living cells, whereas the LDH assay measures the activity of LDH released into the medium from dead cells. In addition, Ca2+ overload may excessively activate the Ca2+/calmodulin-dependent pathways under a variety of pathologic conditions, leading to cellular damage[5]. The viability of the RAW264.7 cells was measured after 48 h of treatment with PM2.5, which showed significant cytotoxicity at high concentrations with a decreased conversion of MTT to the formazan derivative. Compared with the control group, the cell viability was significantly decreased in groups treated with PM2.5 50, 100, 200, and 400 μg/mL (Figure 1A). The decreased cell viability may be correlated with a variety of factors, such as increased cell death or decreased cell proliferation. To clarify the characteristics of PM2.5-induced cytotoxicity, intracellular LDH release and cytosolic [Ca2+]i were determined. We found that PM2.5 triggered significantly higher LDH release from the cytoplasm and increased [Ca2+]i in treated cells than in control cells at a PM2.5 concentration of 100 µg/mL and above, as shown in Figure 1A. Collectively, the elevated LDH leakage, increased [Ca2+]i and decreased MTT metabolic activity induced by PM2.5 at 100 µg/mL and above indicated increased cell death. The flow cytometric apoptosis results showed that PM2.5 noticeably activated apoptosis in a dose-dependent manner (P < 0.01). These results indicated that PM2.5 differentially impacted cell apoptosis. Based on the results of the cell viability assay, cell apoptosis was examined in RAW264.7 cells treated with PM2.5 50, 100, 200, and 400 μg/mL. The results showed that PM2.5 treatment increased apoptosis in RAW264.7 macrophages, as shown. Our results clearly demonstrated that exposure to PM2.5 at a concentration of 100 µg/mL and above could significantly induce ROS generation, decrease cell viability, trigger additional LDH release from the cytoplasm, and increase [Ca2+]i and the percentage of cells in the G2/M phase, which might cause cell damage through oxidative stress.which might cause cell damage through oxidative stress.
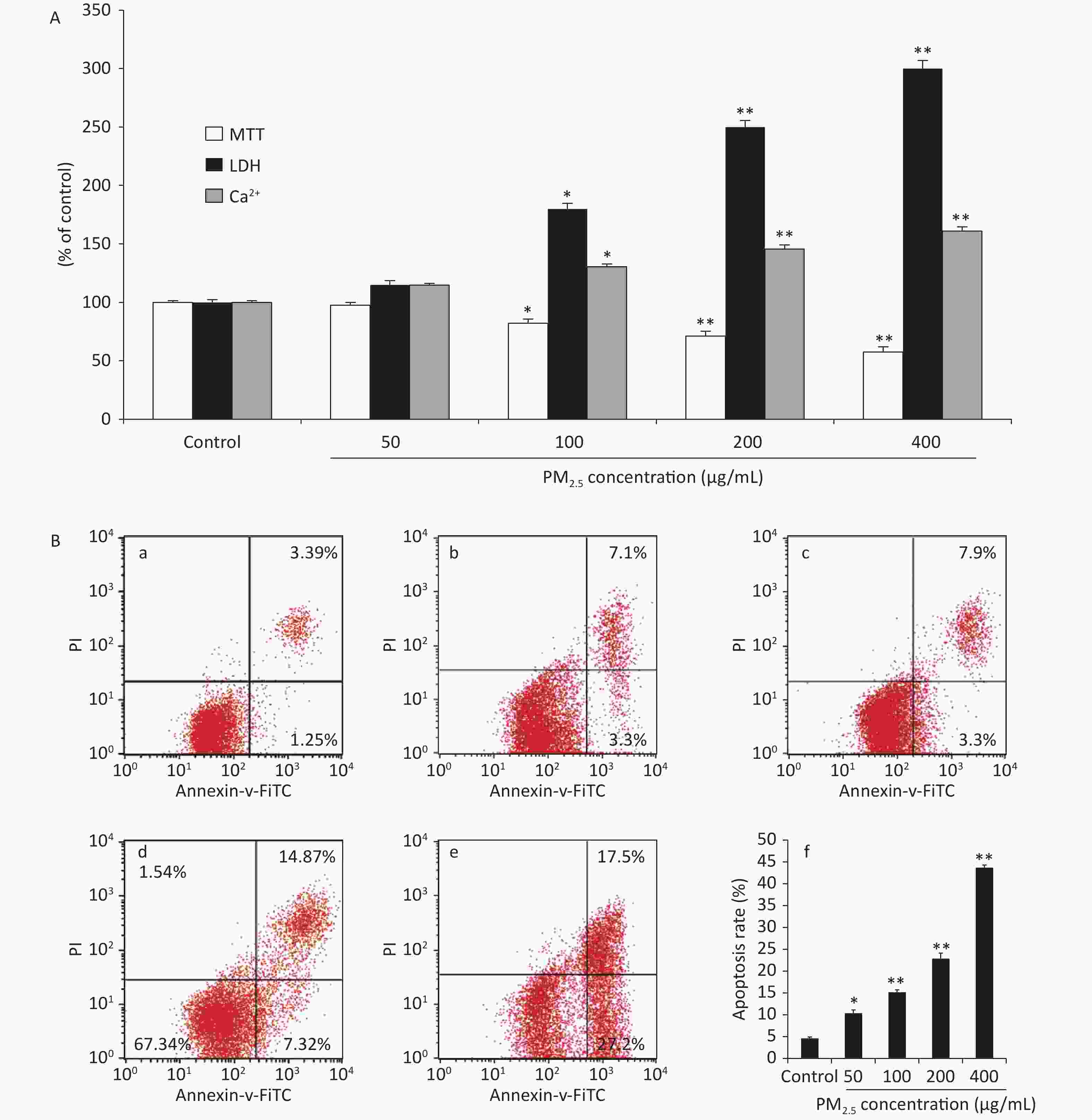
Figure 1. Cytotoxic effects on RAW264.7 cells exposed to PM2.5 at different concentrations for 48 h. (A) MTT assay, LDH leakage and Ca2+ concentration; (B) The effects of PM2.5 on the apoptotic rate based on flow cytometry; (a) Controls, (b) PM2.5 50 μg/mL, (c) PM2.5 100 μg/mL, (d) PM2.5 200 μg/mL, (e) PM2.5 400 μg/mL, (f) Quantification of apoptosis. Values are expressed as the mean ± SEM (n = 6). *P < 0.05, **P < 0.01, compared with control.
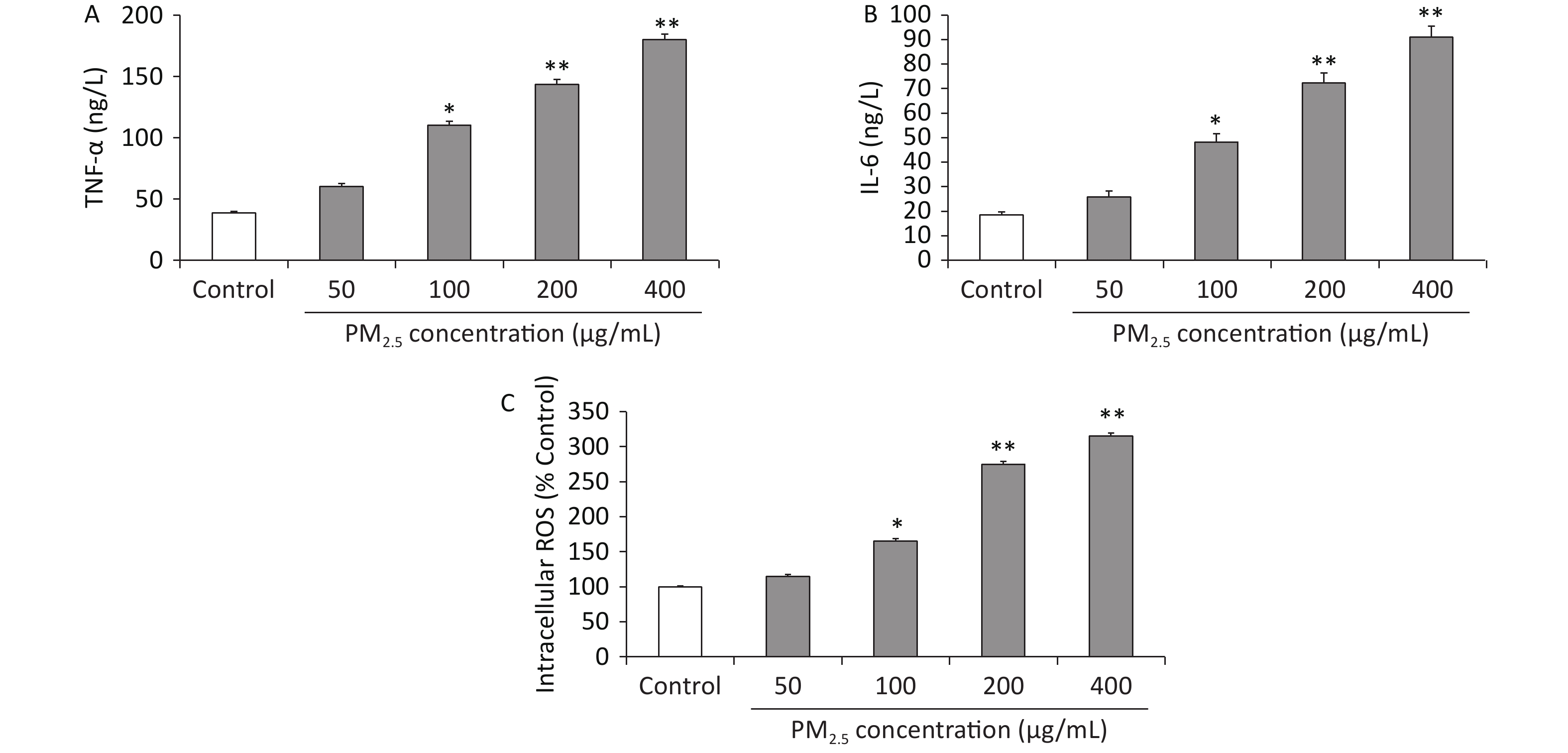
Long-term exposure to PM2.5 can induce excessive reactive oxygen species (ROS) and proinflammatory cytokine production, which are critical for PM2.5-mediated toxicity[1]. Indeed, studies on human subjects and animal models have shown that PM2.5 exposure results in local and systemic inflammation[6]. However, the molecular mechanisms underlying the PM2.5-induced inflammatory response have not been elucidated. Nuclear factor NF-(κB) signaling, which is crucial for inflammation, is activated by numerous factors, such as toll-like receptor (TLR) ligands of lipopolysaccharides (LPS), hypoxia, and tumor necrosis factor (TNF)-α. Furthermore, excessive ROS production can activate the inhibitor of NF-κB (IκB) kinase (IKK) isoforms, including IKKα and IKKβ. These isoforms phosphorylate IκBα to release transcription factors such as NF-kBp65 and subsequently promote its nuclear translocation, leading to the production of proinflammatory cytokines such as interleukin (IL)-6 and IL-8. Hence, the proinflammatory cytokines TNF-α and IL-6 were detected to assess the inflammatory response of macrophages to PM2.5 exposure [7]. On this basis, the concentrations of the proinflammatory cytokines TNF-α and IL-6 in the cell culture medium were analyzed using commercially available enzyme-linked immunosorbent assay (ELISA) kits, and the intracellular ROS level was detected using the oxidation-sensitive fluorescent dye 2ʹ,7ʹ-dichlorodihydrofluorescein. The results showed that PM2.5 at concentrations of 100 µg/mL and above resulted in the elevated release of TNF-α levels compared to controls (P < 0.05, P < 0.01, and P < 0.01). These data indicated that high concentrations of PM2.5 increased TNF-α levels approximately 3-fold that of unexposed controls (Figure 2A). The IL-6 release showed a similar pattern (at a concentration of 100 µg/mL and above approximately 3 times over those with nonexposed controls) to that observed with TNF-α (Figure 2B). The intracellular ROS level showed that PM2.5 at 50 µg/mL and above increased the levels of intracellular ROS, with PM2.5 showing the strongest effects at 400 μg/mL in Figure 2C. These results indicated that exposure to PM2.5 could lead to the production of proinflammatory cytokines.
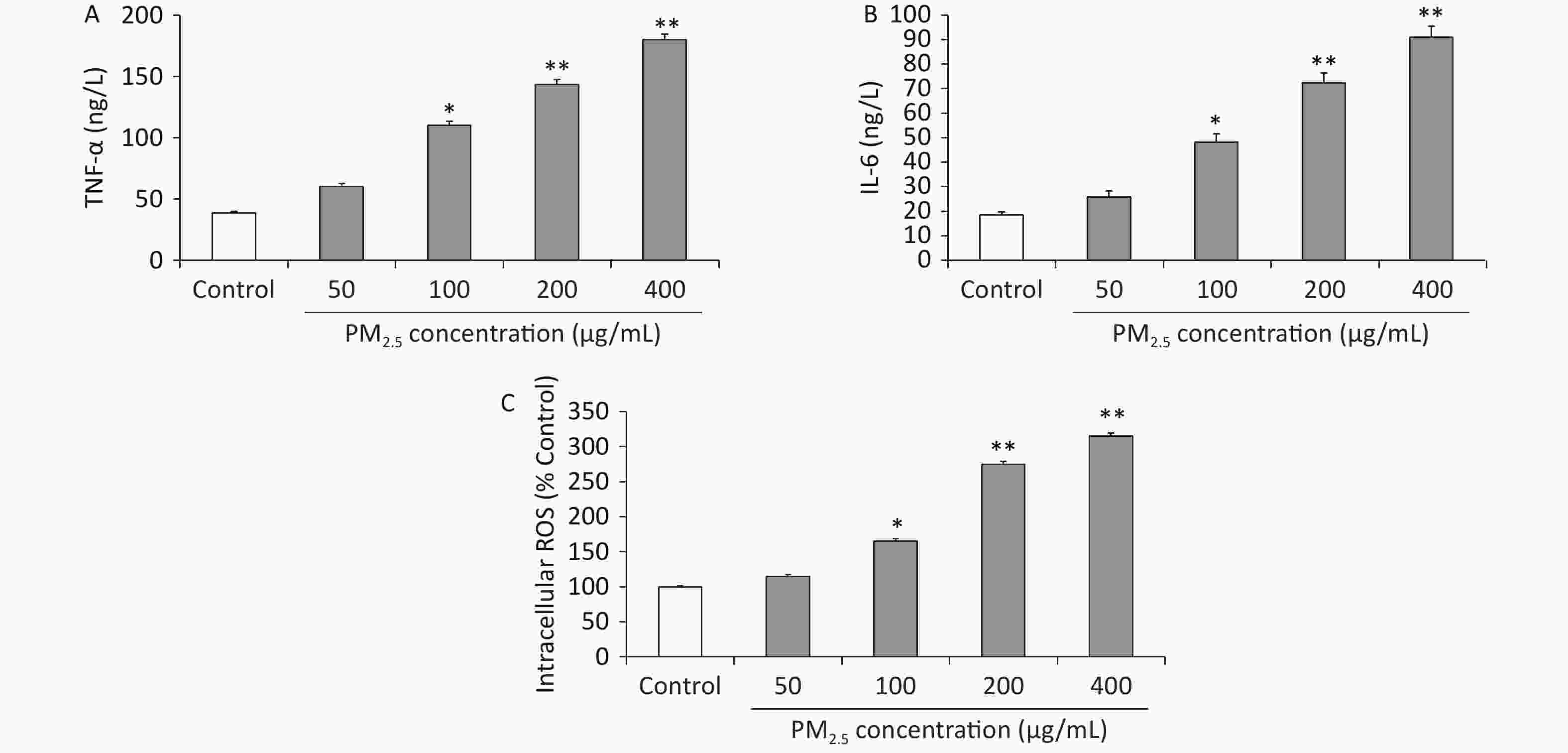
Figure 2. (A) Tumor necrosis alpha (TNF-a) production (ng/L) in RAW 264.7 cells; (B) interleukin-6 (IL-6) production (ng/L) in RAW 264.7 cells; (C) intracellular reactive oxygen species (ROS) levels in RAW264.7 cells. Values are mean ± S.E.M. (n = 6). *P < 0.05, **P < 0.01, compared with control.
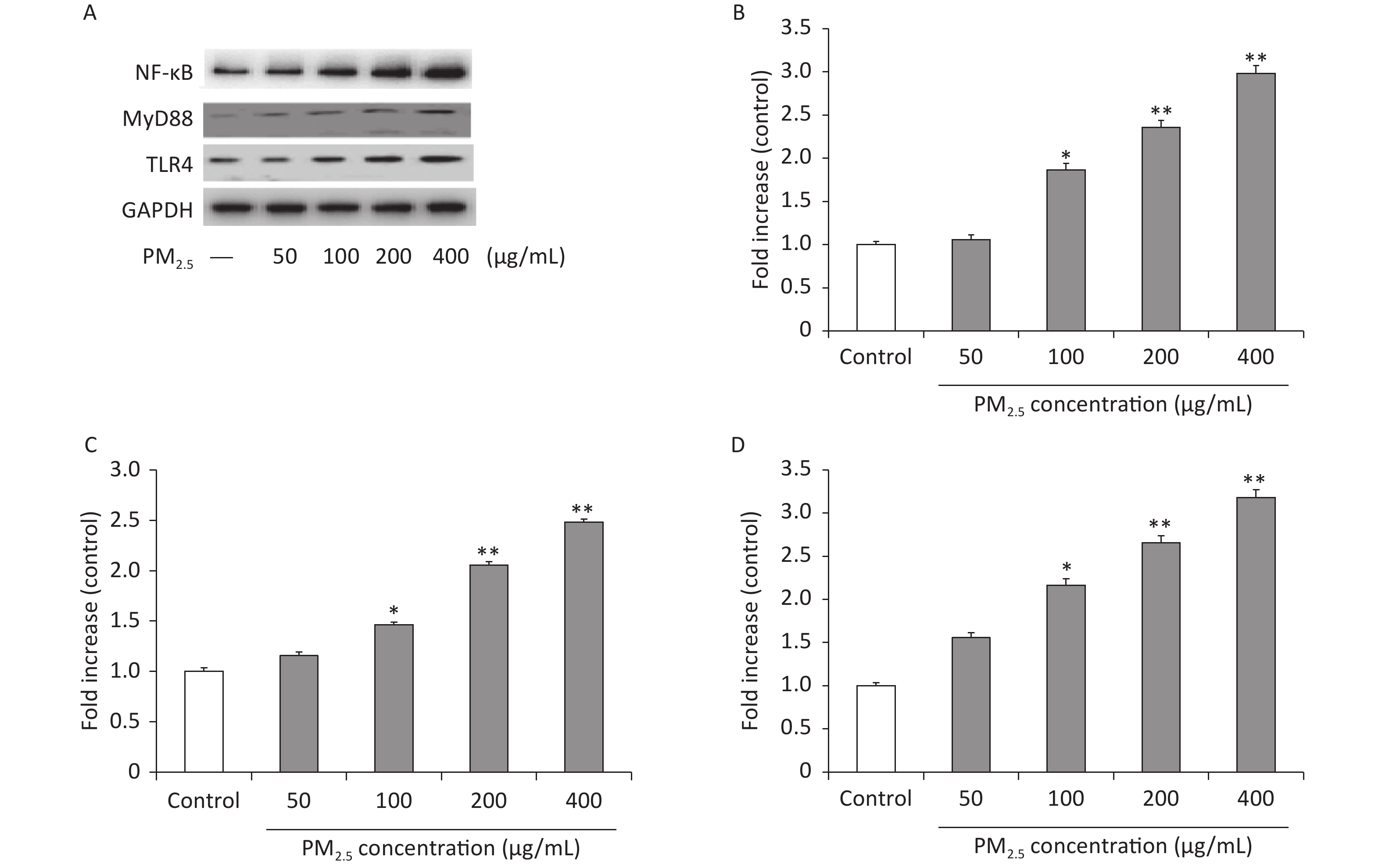
In addition, the inflammatory reaction is a common pathological process of numerous diseases. For example, serious fibrosis development is always associated with the upregulation of inflammatory responses, induced mainly by the activation of the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways [8]. Moreover, the TLR family of proteins plays a fundamental role in pathogen recognition and the activation of innate immunity. The various TLRs exhibit different patterns of expression, and upon activation by stimulation, they mediate immune responses by triggering a cascade of distinct events. TLR4 has been shown to play a key role in the inflammatory response by acting as a receptor for mobilizing NF-κB and promoting the production of inflammatory cytokines[9]. It directly activates downstream signaling pathways by interacting with myeloid differentiation primary response 88 (MyD88), further degrading IκBα and decreasing phosphorylated NF-κB levels [10]. Moreover, in fibrosis, inflammation-related cell infiltration is the key target of antifibrosis research[4]. Based on this knowledge, we investigated whether PM2.5-induced inflammatory responses in mouse macrophages were mediated via activation of the TLR4 and NF-κB signaling pathways. Western blots were performed in Figure 3B to investigate the protein expression levels of NF-κB, which were significantly increased after PM2.5 treatment compared with the controls (P < 0.01), especially at 400 µg/mL, 2.98-fold over those with nonexposed controls. The Western blot analysis in Figure 3C shows that the protein expression levels of MyD88 were significantly increased after PM2.5 treatment compared with the controls (P < 0.01), especially at 400 µg/mL, which was approximately 3 times that of the nonexposed controls. As shown in Figure 3D, compared with the controls, the levels of TLR4 were markedly increased after PM2.5 treatment compared with the controls (P < 0.01), especially at 400 µg/mL, which was approximately three times higher than that of the unexposed control group. The results showed that PM2.5 stimulated the TLR4/NF-κB/MyD88 signaling pathway, which enhanced phosphorylated NF-κB levels and proinflammatory cytokine release and potentially accelerated lung injury.
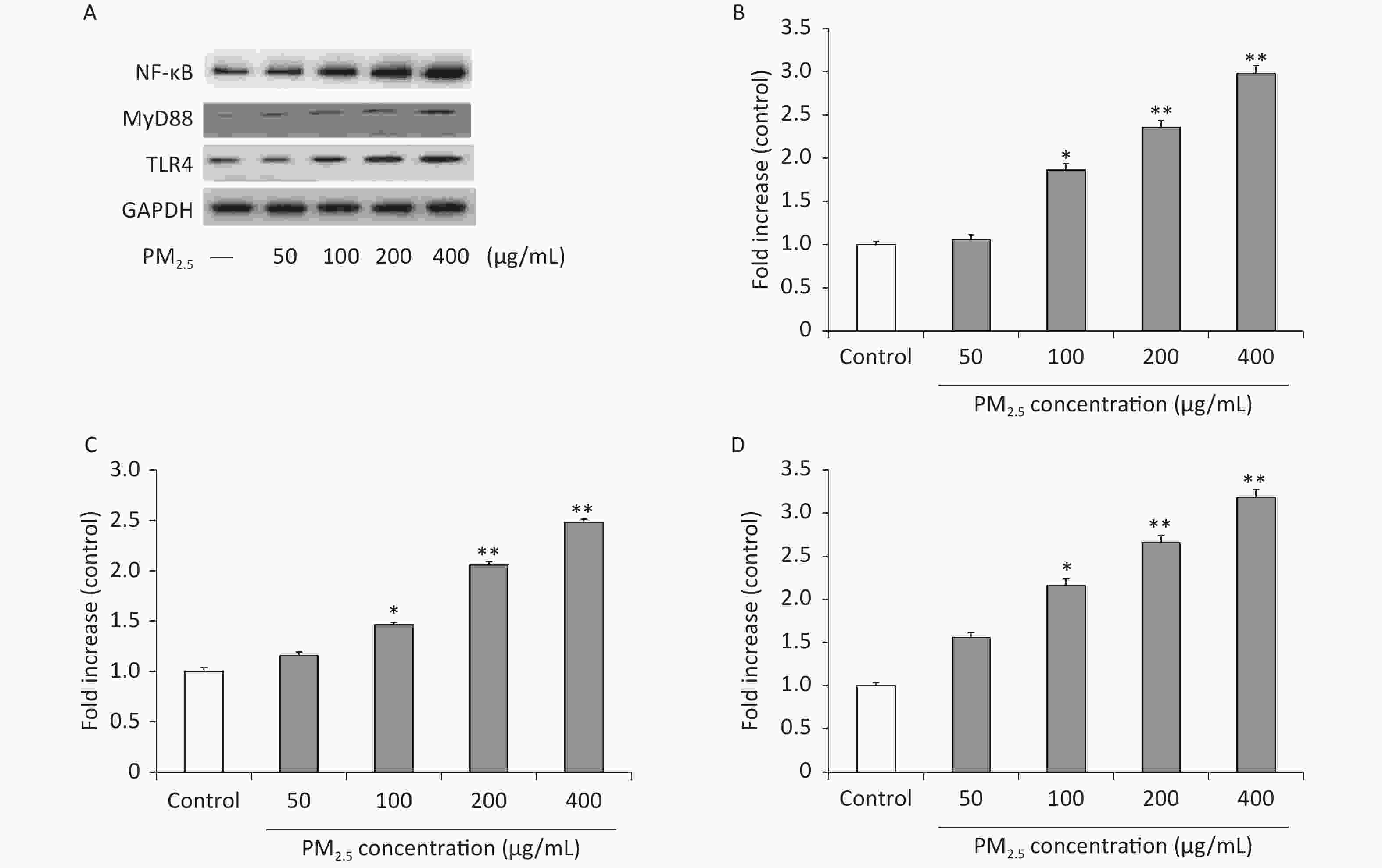
Figure 3. PM2.5 regulates the TLR4/MyD88/NF-κB pathway in RAW 264.7 cells. Western blot (A) and analysis of NF-κB (B), MyD88 (C), TLR4 (D). Values are in the form of means ± SEM (n = 6). Values are mean ± SEM (n = 6). *P < 0.05, **P < 0.01, compared with control.
In the present study, we demonstrated that PM2.5 induced early response ROS generation, decreased cell viability, triggered cytoplasmic LDH release, and increased [Ca2+]i and the percentage of G2/M cells, possibly causing cell damage through oxidative stress. Additionally, high concentrations of PM2.5 (200 and 400 μg/mL) significantly increased IL-6 and TNF-α levels and promoted NF-κB, MyD88, and TLR4 protein expression compared to unexposed controls. These findings show that atmospheric environmental PM2.5 in Changchun city causes inflammation and oxidative stress in macrophages via TLR4/NF-κB/MyD88 signaling pathways. Our findings provide new insights into the molecular mechanisms underlying PM2.5 toxicities and may facilitate the development of novel preventative strategies.
Conflicts of Interest
ZHENG Mei Zhu, LU Yao, Lu Ting Ting, QIN Peng, LI Yu Qiu, and SHI Dong Fang, declare that they have no conflicts of interest.
Ethical Statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Author Contributions
ZHENG Mei Zhu designed the research; LU Ting Ting and LU Yao performed the research; ZHENG Mei Zhu, Qin Peng and SHI Dong Fang analyzed the data; ZHENG Mei Zhu and LU Yao wrote the paper; LI Yu Qiu reviewed the manuscript.
doi: 10.3967/bes2023.055
Fine Particulate Matter Exposure Induces Toxicity by Regulating Nuclear Factor-κB/toll-like Receptor 4/myeloid Differentiation Primary Response Signaling Pathways in RAW264.7 Cells
-
&These authors contributed equally to this work.
注释: -
Figure 1. Cytotoxic effects on RAW264.7 cells exposed to PM2.5 at different concentrations for 48 h. (A) MTT assay, LDH leakage and Ca2+ concentration; (B) The effects of PM2.5 on the apoptotic rate based on flow cytometry; (a) Controls, (b) PM2.5 50 μg/mL, (c) PM2.5 100 μg/mL, (d) PM2.5 200 μg/mL, (e) PM2.5 400 μg/mL, (f) Quantification of apoptosis. Values are expressed as the mean ± SEM (n = 6). *P < 0.05, **P < 0.01, compared with control.
Figure 2. (A) Tumor necrosis alpha (TNF-a) production (ng/L) in RAW 264.7 cells; (B) interleukin-6 (IL-6) production (ng/L) in RAW 264.7 cells; (C) intracellular reactive oxygen species (ROS) levels in RAW264.7 cells. Values are mean ± S.E.M. (n = 6). *P < 0.05, **P < 0.01, compared with control.
-
[1] Jin XT, Su RJ, Li RJ, et al. Amelioration of particulate matter-induced oxidative damage by vitamin c and quercetin in human bronchial epithelial cells. Chemosphere, 2016; 144, 459−66. doi: 10.1016/j.chemosphere.2015.09.023 [2] Zhou ZX, Liu YH, Duan FK, et al. Transcriptomic analyses of the biological effects of airborne PM2.5 exposure on human bronchial epithelial cells. PLoS One, 2015; 10, e0138267. doi: 10.1371/journal.pone.0138267 [3] Lin HW, Chen M, Gao YJ, et al. Tussilagone protects acute lung injury from PM2.5 via alleviating Hif-1α/NF-κB-mediated inflammatory response. Environ Toxicol, 2022; 37, 1198−210. doi: 10.1002/tox.23476 [4] Geng J, Liu HY, Ge PB, et al. PM2.5 promotes plaque vulnerability at different stages of atherosclerosis and the formation of foam cells via TLR4/MyD88/NFκB pathway. Ecotoxicol Environ Saf, 2019; 176, 76−84. doi: 10.1016/j.ecoenv.2019.03.068 [5] Ma P, Yan B, Zeng Q, et al. Oral exposure of Kunming mice to diisononyl phthalate induces hepatic and renal tissue injury through the accumulation of ROS. Protective effect of melatonin. Food Chem Toxicol, 2014; 68, 247−56. doi: 10.1016/j.fct.2014.03.027 [6] Ostro B, Malig B, Broadwin R, et al. Chronic PM2.5 exposure and inflammation: determining sensitive subgroups in mid-life women. Environ Res, 2014; 132, 168−75. doi: 10.1016/j.envres.2014.03.042 [7] Lyu Y, Zhou JT, Li JJ, et al. Alterations of IL-1beta and TNF-alpha expression in RAW264.7 cell damage induced by two samples of PM2.5 with different compositions. Sci Prog, 2022; 105, 368504221113709. [8] Kar S, Ukil A, Das PK. Cystatin cures visceral leishmaniasis by NF-κB-mediated proinflammatory response through co-ordination of TLR/MyD88 signaling with p105-Tpl2-ERK pathway. Eur J Immunol, 2011; 41, 116−27. doi: 10.1002/eji.201040533 [9] Wang BX, Sun T, Sun L, et al. Amygdalin attenuates PM2.5-induced human umbilical vein endothelial cell injury via the TLR4/NF-κB and Bcl-2/Bax signaling pathways. Acta Biochim Biophys Sin (Shanghai), 2022; 54, 1476−85. doi: 10.3724/abbs.2022136 [10] Zhang J, Chen XW, Li H, et al. Selenium-enriched soybean peptides pretreatment attenuates lung injury in mice induced by fine particulate matters (PM2.5) through inhibition of TLR4/NF-κB/IκBα signaling pathway and inflammasome generation. Food Funct, 2022; 13, 9459−69. doi: 10.1039/D2FO01585D -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1089
- HTML全文浏览量: 497
- PDF下载量: 40
- 被引次数: 0

 Quick Links
Quick Links