

 下载:
下载:

-
Antimony (Sb) can impair respiratory and cardiovascular function as a result of natural or occupational exposure[1]. Antimony was found in human brain tissues more than 40 years ago[2]. Recently, Tanu et al.[3] evaluated antimony-associated neurotoxicity in intraperitoneal-injected mice by assessing its accumulation in the brain accumulation and measuring cognitive function using the Morris water maze. Subsequently, our group investigated the cellular outcomes and molecular mechanism of antimony-related neurotoxicity. Wang et al.[4] first reported that antimony triggered autophagic death in neurons by inhibiting the reactive oxygen species (ROS)-dependent mammalian target of rapamycin signaling pathway. Based on this evidence, antimony was characterized as a novel neurotoxicant. In addition to neurons, the central nervous system (CNS) contains gliocytes, astrocytes, oligodendrocytes, and microglia, all of which potentially play important roles in metal-associated neurotoxicity. For example, arsenic trioxide triggers a microglial inflammatory response, resulting in neuronal apoptosis[5]. Cadmium accumulated in cultured astrocytes and stimulated inflammatory factor release via the mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NF-κB) pathways[6]. Sub-toxic concentrations of heavy metals led to dysfunctional oligodendrocytes[7]. Therefore, brain cells other than neurons may respond to antimony exposure. In order to clarify the mechanism(s) of antimony-mediated neurotoxicity, it is important to investigate the effects of antimony on these cell types.
Astrocytes are abundant, conserved CNS cells that maintain normal CNS function, support the blood-brain barrier, and modulate synaptic activity[8, 9]. Astrocytes also play roles in the progression of various neurological diseases[10]. Astrocyte reactivity is an early feature of Alzheimer’s disease (AD), potentially providing a target for preclinical diagnosis and treatment[11]. Astrocytes are activated by numerous environmental stimuli[12, 13]. Reactive astrocytes are thought to be characterized by elevated expression of glial fibrillary acidic protein (GFAP), increased cell proliferation, and massive release of inflammatory factors [e.g., tumor necrosis factor-alpha (TNF-α), interleukin 1 beta (IL-1β), and interleukin 6 (IL-6)]. The impact of reactive astrogliosis in neurological diseases is complex; it appears to be beneficial in the initial stage of many neurodegenerative diseases by taking up and clearing β-amyloid protein (Aβ) from the brain[14]. Reactive astrocytes surrounding Aβ plaques contribute to the local inflammatory response and are therefore involved in AD development.
Astrocyte activation is a complex process regulated by many critical signaling pathways, including NF-κB. NF-κB is an important transcription factor that participates in different cellular pathways involving apoptosis, oxidative stress, and inflammation[15]. In astrocytes, NF-κB signaling contributes to pro-inflammatory responses following injury[16]. Moreover, NF-κB acts as a signaling hub that collects upstream signals to mediate astrocyte activation. For example, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) promoted the inflammatory activation of astrocytes via the TGF-β-activated kinase 1 (TAK1)–NF-κB cascade, while blockage of TAK1 effectively attenuated TCDD-induced NF-κB activation[17]. In addition, manganese-exposed astrocytes released inflammatory mediators in a NF-κB-dependent manner, leading to neuron death[18].
In this study, we investigated whether antimony exposure stimulates astrocyte activation in vivo and in vitro. We found that low concentrations of antimony triggered astrocyte activation via NF-κB signaling. Our study provides new evidence of antimony-associated neurotoxicity, supporting the idea that antimony is a novel nerve poison.
-
The chemical regents and antibodies were purchased as follows: potassium antimonyl tartrate trihydrate (Sigma, 60063); Hoechst (Sigma, 94403); TAK1 inhibitor (TargetMol, T4264); BAY11-7082 (MCE, HY-13453); anti-β-actin (Santa Cruz Biotechnology, sc-47778); anti-p65 (Abcam, ab16502); anti-p-p65 (Abcam, ab86299); anti-GFAP (CST, 12389S); anti-iNOS (GeneTex, GTX130246); anti-Lamin A/C (Proteintech, 10298-1-AP); anti-Tubulin (Proteintech, 10068-1-AP); anti-TAK1 (Abcam, ab109526); anti-p-TAK1 (Abcam, ab109404); and anti-Cyclin D1 (Cell Signaling Technology, 2978).
-
We performed mouse experiments in accordance with the ethical guidelines from the Ethics Committee of Laboratory Animal Care and Welfare (Nantong University School of Medicine). Thirty ICR male mice (weight ≥ 27 g, 6 weeks of age) were provided by the Experimental Animal Center of Nantong University. After acclimatization to a 12/12 h dark/light cycle for one week, mice were randomly divided into three groups. In each group, 100 μL antimony (0, 10, and 20 mg/kg) dissolved in 0.9% saline solution was injected intraperitoneally. This dose was used to explore antimony-associated neurotoxicological mechanism because it had a significant toxic effect on neurobehavioral change in mice under present conditions[3]. Moreover, during treatment of schistosomiasis, the patients were exposed to antimony potassium tartrate at a higher dose (2.5 g)[19]. More importantly, in the human brain, the antimony concentration is ≥ 2.5 × 10−6–170.7 × 10−6 g/g[2]. Antimony treatment was performed as follows. Antimony was injected intraperitoneally on the first, third, and fifth days of every week for eight weeks, and the mice were sacrificed on the first day of the ninth week. All mice were anesthetized by intraperitoneal injection of 7% chloral hydrate (5 mL/kg). Mice were perfused intracardially with saline and 4% paraformaldehyde for paraffin embedding. Moreover, cerebral cortices were collected and frozen at −80 °C for immunoblot analysis.
-
C6 cells were cultured aseptically according to conventional culture methods. F12K (Gibco) medium was supplemented with 2.5% fetal bovine serum (FBS, Gibco) and 15% horse serum (HyClone, SH30074.02) and cells were grown at 37 °C and with 5% CO2. Antimony was dissolved in 0.9% normal saline, and C6 cells were seeded for 24 h and then exposed to an Antimony concentration gradient (0, 0.625, 1.25, 2.5, 5 μmol/L) for another 24 h. Moreover, cells were exposed to 2.5 μmol/L antimony for the time gradient assay (0, 3, 6, 12, 24 h). C6 cells were pretreated with TAK1 and p65 inhibitors for 1 h and exposed to 2.5 μmol/L for 24 h, then protein or RNA were collected and stored at −80 °C.
-
C6 cells were seeded in 96-well plates (≥ 10,000 cells/well) and exposed to varying antimony concentrations for 24 h. After exposure, 10 μL of the reaction solution were added to each well according to the instructions of the CCK8 kit (DOJINDO, Japan, CK04) as described by Zhao et al.[20]. The optical density (OD) value at 450 nm was detected with a microplate reader 2 h later (Thermo Scientific, Varips Kan Flash).
-
Total protein was extracted from mouse brain tissue and C6 cells, and the protein concentrations were measured using the BCA kit (Beyotime, P0009). An equal amount of protein per sample was subjected to electrophoresis (10% SDS-PAGE) to separate the proteins, which were then transferred to a PVDF membrane. The membrane was blocked in a TBST (Tris-buffered saline) buffer containing 3% BSA (bovine serum albumin) for at least 2 h at room temperature, and then incubated with primary antibody at 4 °C overnight. The corresponding secondary antibodies were then incubated with the membrane for 1 h, and finally, protein bands were detected using an enhanced chemiluminescence system (ECL; Millipore Corporation, Billerica, P90720).
-
C6 cells exposed to antimony were collected, their cytoplasmic proteins were extracted with cytoplasmic protein extractants A and B according to the instructions for the Nuclear Separation kit (Beyotime, P0027)[21], and the nuclear protein was extracted with the nuclear protein extraction reagents. Finally, the expression of p65 protein was detected by immunoblotting.
-
C6 cells on coverslips were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 15 min at 4 °C, washed another three times with PBS and permeabilized with 0.2% Triton X-100 for 15 min at 4 °C. Next, the cells were blocked with goat serum for 2 h at room temperature. Then, the slides were incubated with primary anti-p65 antibody (diluted 1:200 in PBS) at 4 °C overnight. The slides were then washed three times with PBS and incubated with the Alexa Fluor 488-conjugated goat anti-rabbit antibody (Invitrogen, R37116) for 1 h followed by Hoechst treatment for 15 min. Cells were observed using a fluorescence microscope (Leica, Microsystems, GmbH, Germany). For fluorescence excitation, an Ar/UV laser was used for Hoechst, and an Ar/Blue laser was used for p65.
-
Paraffin-embedded brain tissue slices (5 μm thickness) were cut in the coronal plane and subjected to immunohistochemistry to detect the expression of GFAP as described by Piccolini et al.[22]. Embedded sections were deparaffinized in xylene and rehydrated through a series of graded alcohol treatments. Then, slices were boiled in 10 mmol/L citrate buffer for 30 min for antigen retrieval. Endogenous peroxidation was blocked with 3% H2O2 for 30 min, then slices were incubated with 5% BSA for 2 h at room temperature in order to block non-specific antigen binding sites. Then, the slices were incubated with GFAP antibody (diluted 1:200 in PBS) at 4 °C overnight, and then incubated with the HRP-conjugated secondary antibody. Sections were washed in PBS three times and finally stained with DAB and hematoxylin. Sections were dehydrated in ethanol, cleared in xylene, and mounted with neutral balsam mounting medium. Slices were imaged using a micro Oscilloscope (Leica, Microsystems, GmbH, Germany) for visualization.
-
Following the kit instructions, total RNA was extracted from C6 cells using Trizol reagent (TakaRa, 9109) and reversed-transcribed to generate cDNA. For RT-PCR, Taq DNA polymerase and specific primers were used. The PCR products of each sample were analyzed by electrophoresis in a 2% agarose gel. The intensity of resulting bands was measured by densitometry and then normalized to the corresponding GAPDH bands. For qRT-PCR, PCR analysis was performed in a 10 μL reaction using SYBR GREEN PCR Master Mix (Applied Biosystems). We performed qRT-PCR to measure gene expression, and GAPDH was chosen as a reference and was calibrated on untreated cells. The delta-delta Ct (
ΔΔCT) method was applied for relative quantification. The corresponding primers are shown in Table 1. Table 1. Oligonucleotide sequences used in PCR
Gene name Primer name Sequence (5’–3’) RAT-GAPDH GAPDH-F TGATGACATCAAGAAGGTGGTGAAG GAPDH-R TCCTTGGAGGCCATGTGGGCCAT RAT-iNOS iNOS-F GCTTTGTGCGGAGTGTCAGT iNOS-R CCTCCTTTGAGCCCTCTGTG RAT-GFAP GFAP-F GGCGCTCAATGCTGGCTTCA GFAP-R TCTGCCTCCAGCCTCAGGTT RAT-IL-6 IL-6-F GAAAGTCAACTCCATCTGCC IL-6-R CATAGCACACTAGGTTTGCC RAT- IL-1β IL-1β-F TCAGGAAGGCAGTGTCACTCATTG IL-1β-R ACACACTAGCAGGTCGTCATCATC RAT-TNF-α TNF-α-F GCCGATTTGCCATTTCAT TNF-α-R CAGTCGCCTCACAGAGCAA -
Quantitative results are presented as the mean ± standard deviation (SD) from at least three independent experiments. Then, analysis of variance (ANOVA) or Student’s t-test were used for statistical analysis. A P value ≤ 0.05 was considered significant.
-
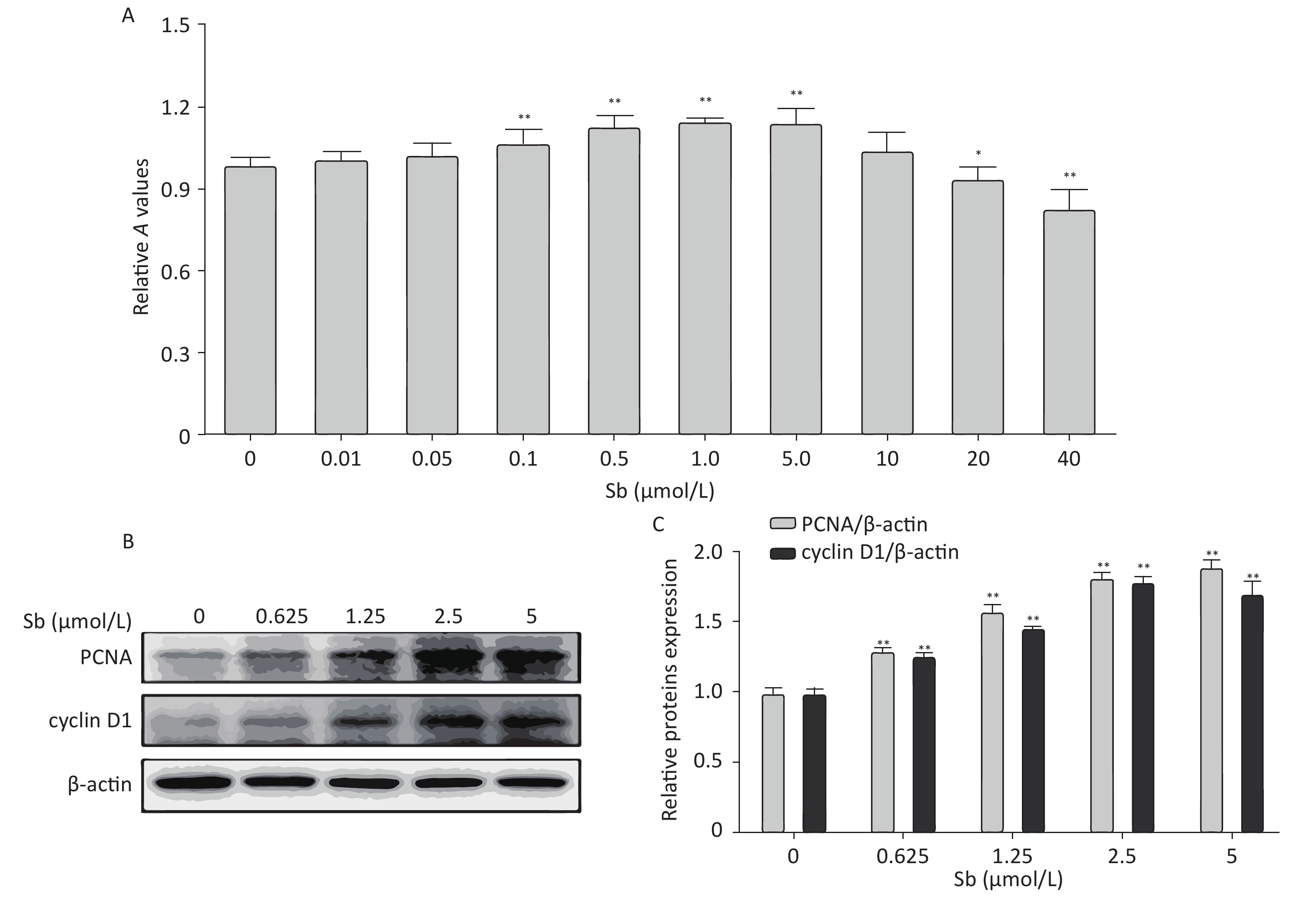
Cell proliferation is an important factor of reactive astrocytes and is the pathological basis of several astrocyte diseases. Therefore, we exposed C6 cells to different concentrations of antimony (0, 0.01, 0.05, 0.1, 1, 5, 10, 20, and 40 μmol/L) for 24 h, and examined cell proliferation using CCK8 assays. Exposure to 0.1–5 μmol/L antimony dramatically promoted cell proliferation in a dose-dependent manner, while exposure to 20–40 μmol/L antimony significantly reduced the CCK8 value (Figure 1A). Therefore, exposure to low concentrations of antimony promotes the proliferation of C6 cells, while high antimony concentrations lead to cell damage.
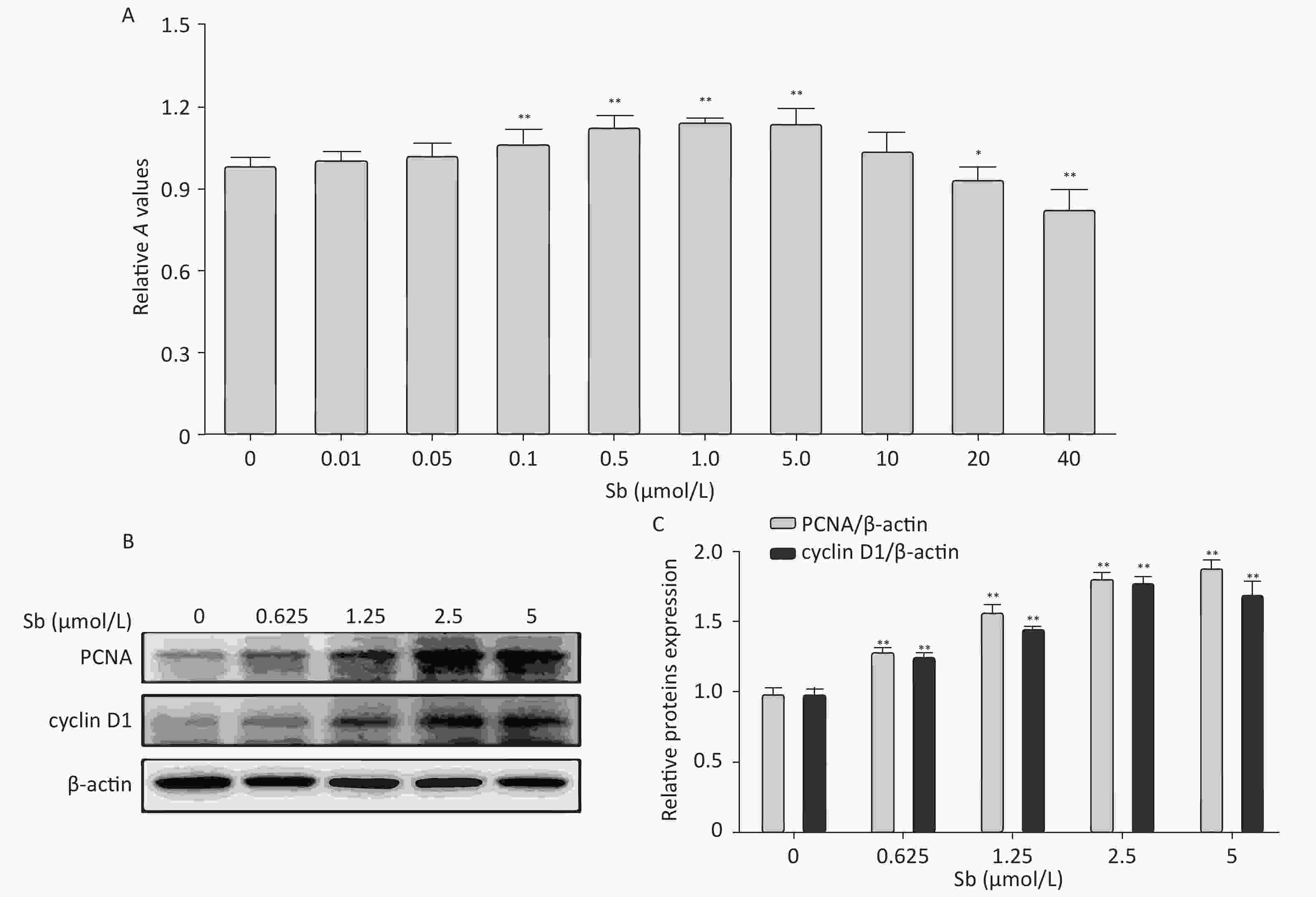
Figure 1. Antimony promotes astrocyte proliferation. (A) C6 cells were exposed to antimony at different concentrations (0, 0.01, 0.05, 0.1, 1, 5, 10, 20, 40 μmol/L) for 24 h, and optical density A value of CCK8 regents was used to assess cell proliferation and cytotoxicity. (B) C6 cells were exposed to the indicated antimony concentrations for 24 h, and expression levels of PCNA and cyclin D1 proteins were detected by western blot. (C) The levels of PCNA and cyclin D1 proteins were quantified by normalization to β-actin. Data are presented as mean ± SD, **P < 0.01, *P < 0.05.
Based on these results, we exposed C6 cells to 0, 0.625, 1.25, 2.5, and 5 μmol/L antimony, and detected two classic cell proliferation markers by immunoblotting. Cyclin D1 is a nuclear protein required for G1 cell cycle progression[23]. Proliferating cell nuclear antigen (PCNA) is an endogenous marker of cell proliferation, which plays an important role in initiating cell proliferation and is thus a good indicator of cell proliferation[24]. Exposure to antimony significantly upregulated cyclin D1 and PCNA expression in C6 cells in a concentration-dependent manner (Figure 1B, 1C). Collectively, these data indicate that antimony promotes C6 cell proliferation in a concentration-dependent manner at low doses, suggesting that astrocytes are activated after antimony exposure.
-
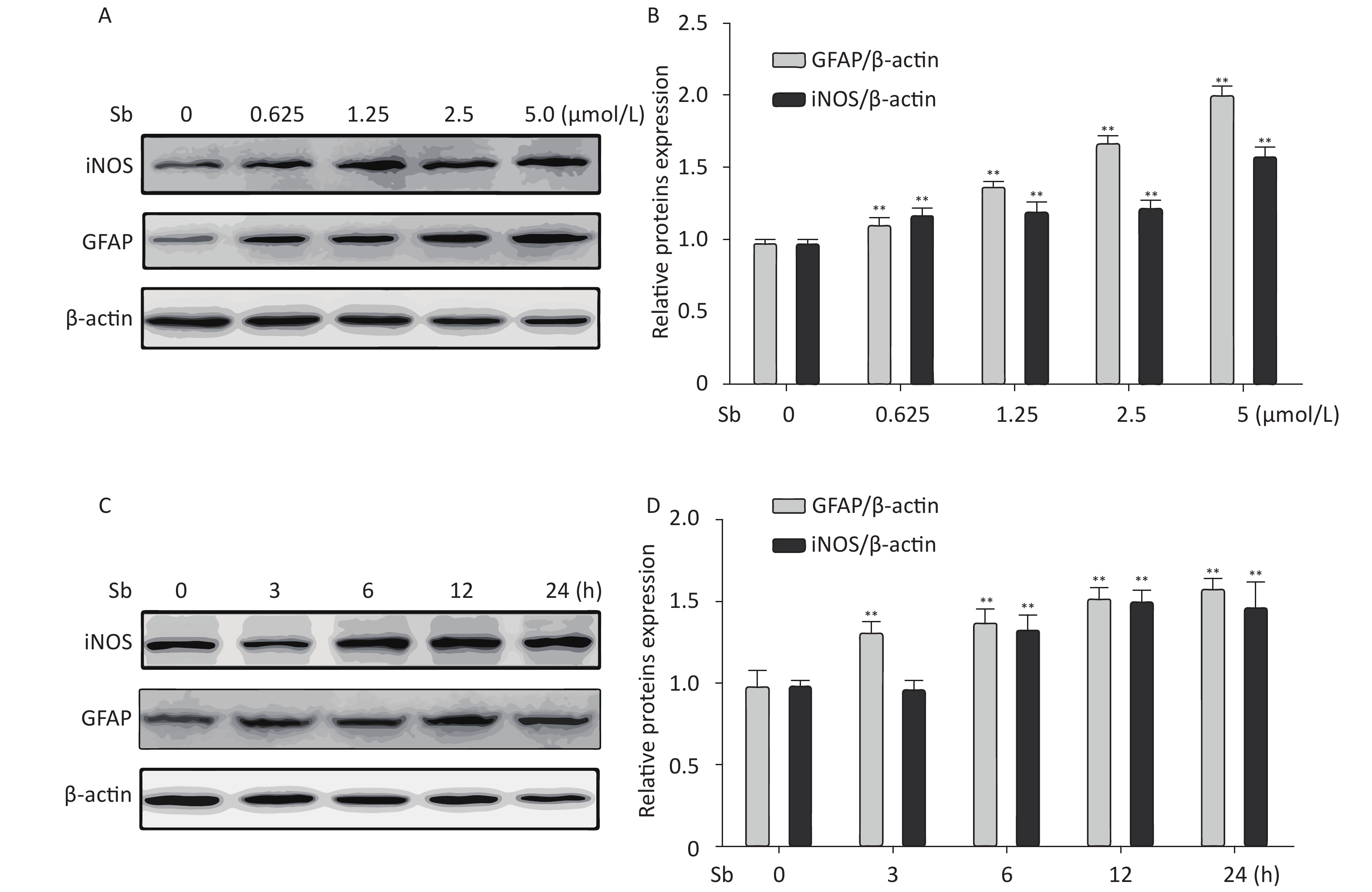
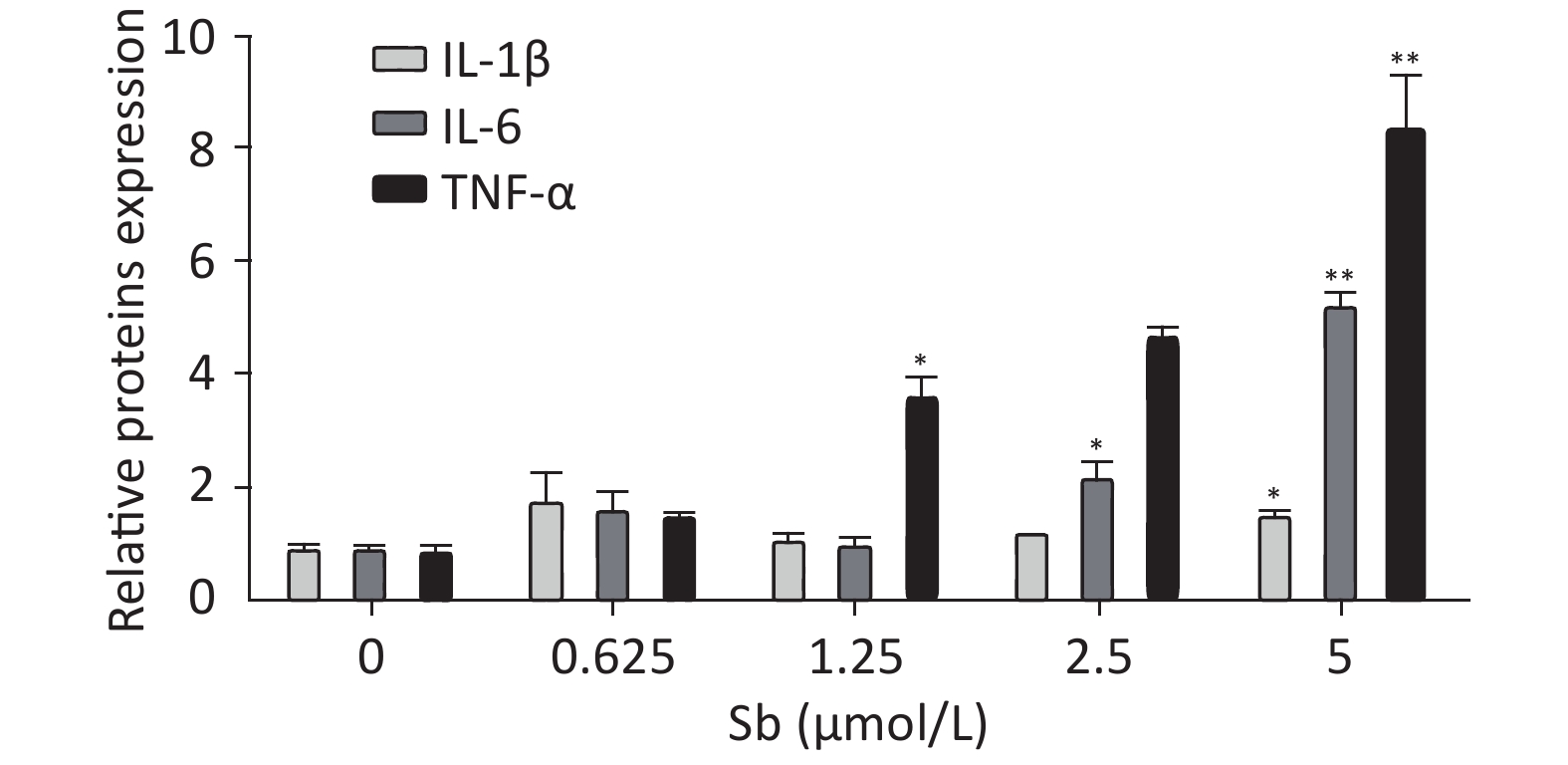
To validate the antimony -induced astrocyte activation results, we established an in vitro model using antimony-exposed C6 cells. C6 cells were exposed to different concentrations of antimony for 24 h and then the expression of GFAP and inducible nitric oxide synthase (iNOS) protein was detected by western blotting. Antimony exposure gradually increased the expression of GFAP and iNOS proteins in a concentration-dependent manner (Figure 2A, 2B). Exposure to 0.625 μmol/L antimony markedly activated C6 cells. We also treated C6 cells with 2.5 μmol/L antimony for varying durations (0, 3, 6, 12, and 24 h) and found that GFAP was significantly increased after 3 h, while iNOS was upregulated after 6 h in C6 cells (Figure 2C, 2D). Furthermore, we found that the expression of IL-1β, IL-6, and TNF-α increased gradually following antimony exposure at varying concentrations (Figure 3), further indicating astrocyte activation.
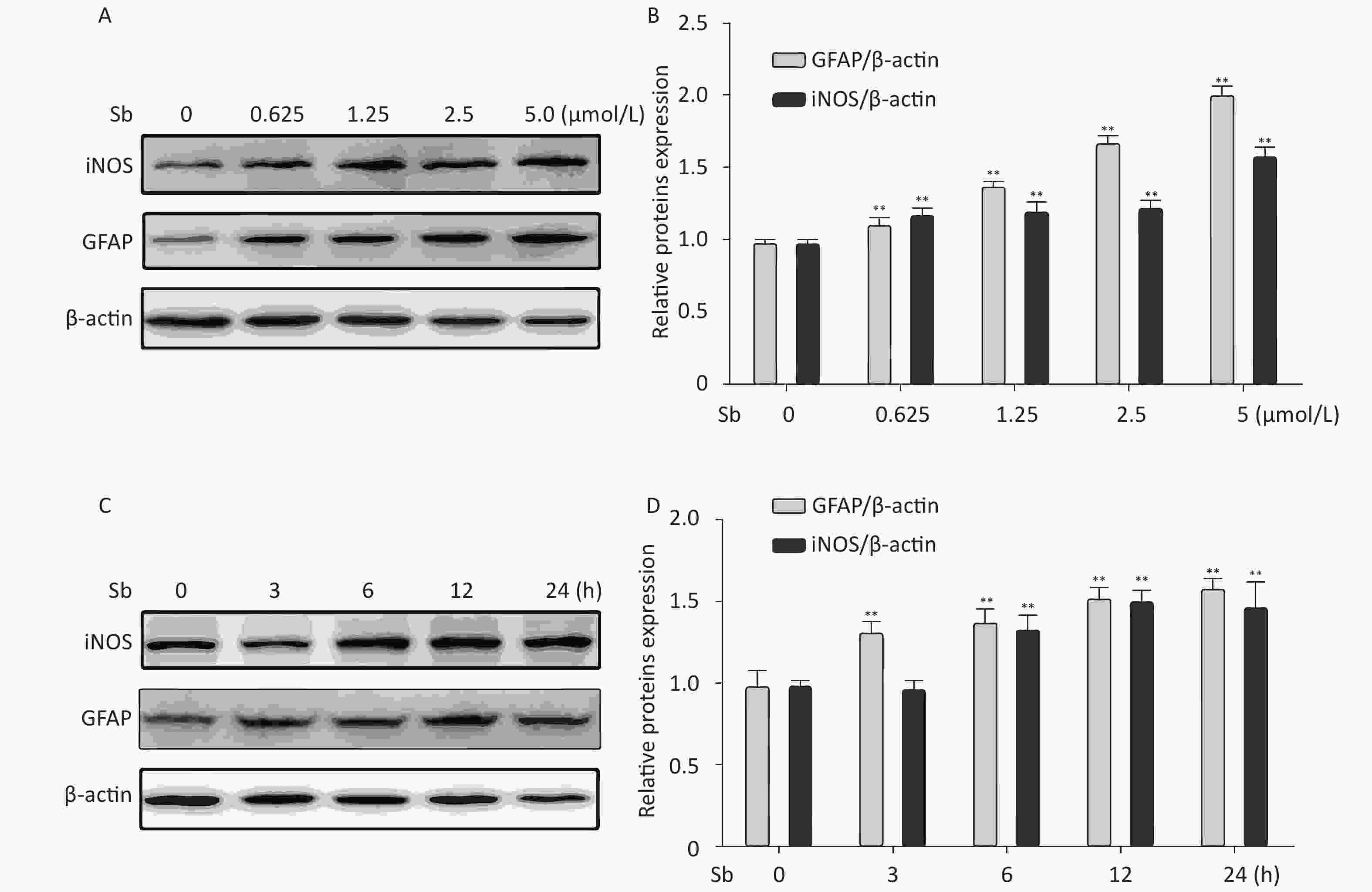
Figure 2. Antimony induces astrocyte activation in vitro. (A) C6 cells were exposed to varying concentrations of antimony for 24 h. The expression levels of GFAP and iNOS proteins were measured by immunoblotting. (B) Quantitative analysis of protein expression relative to β-actin expression for (A). (C) C6 cells were exposed to antimony at 2.5 μmol/L, and the proteins expression levels were detected at different times (0, 3, 6, 12, 24 h) by immunoblotting. (D) The levels of GFAP and iNOS proteins were quantified by normalization to β-actin. Data are presented as mean ± SD, **P < 0.01.
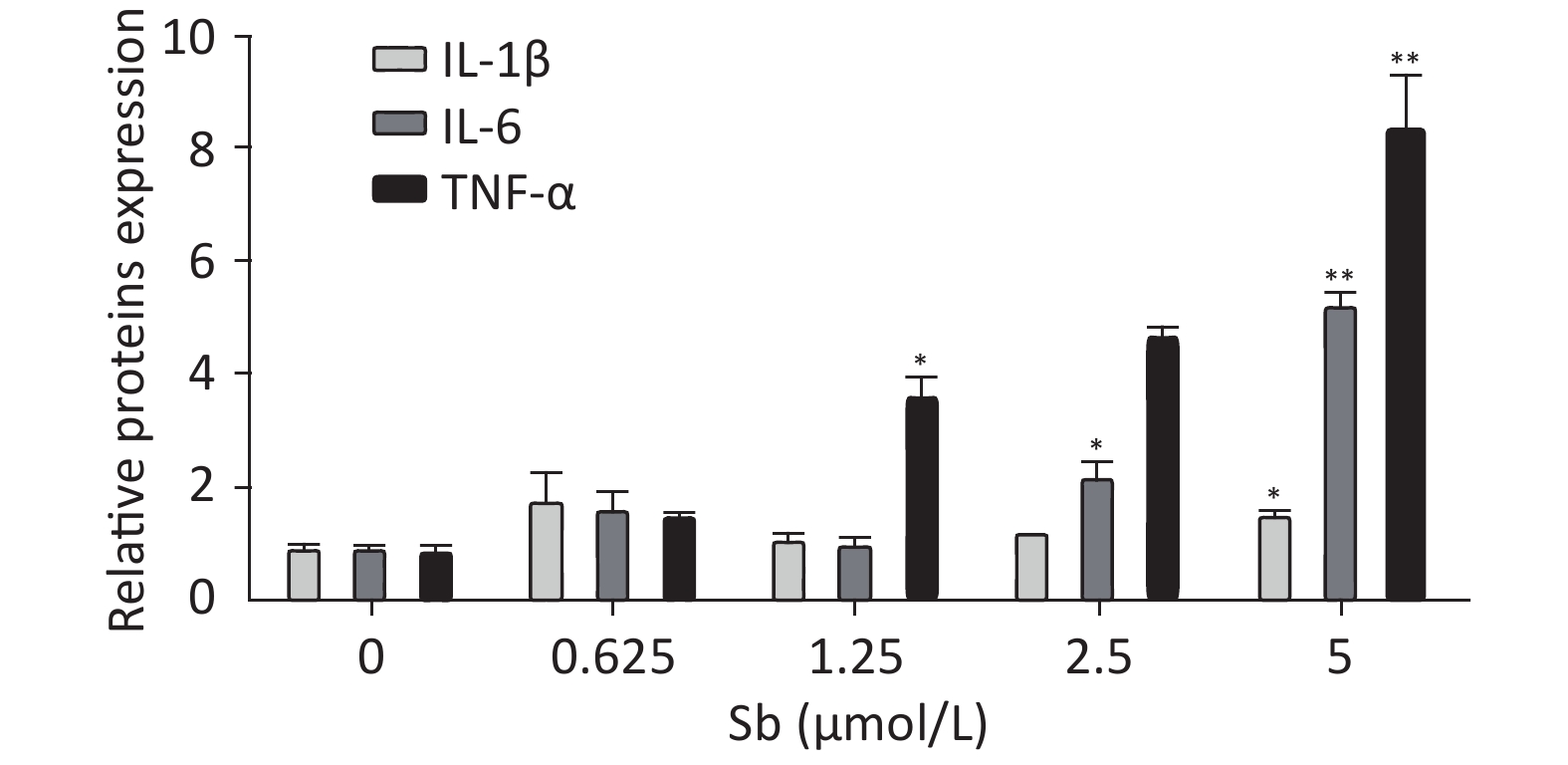
Figure 3. Antimony upregulates inflammatory factors in C6 cells. C6 cells were exposed to antimony at the indicated doses, and the levels of IL-1β, IL-6, and TNF-α mRNAs were quantified by qRT-PCR. Data are presented as mean ± SD, **P < 0.01, *P < 0.05.
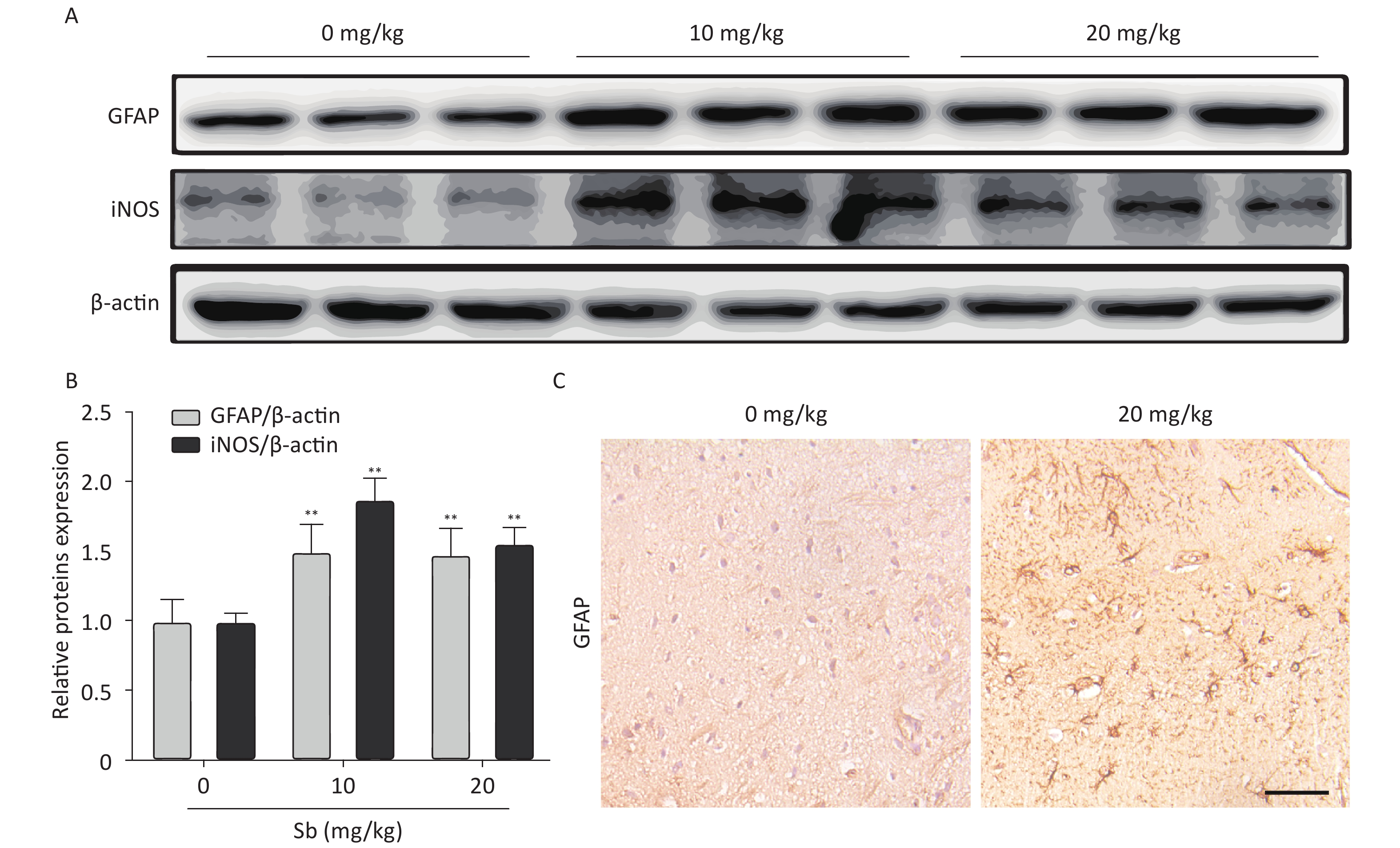
To confirm that antimony can stimulate astrocyte activation in vivo, we constructed an antimony-exposed mouse model. After exposure, we used immunoblotting to detect GFAP and iNOS expression in brain tissues. Compared with controls, the expression of GFAP and iNOS proteins was significantly increased in the brain tissue of mice exposed to 10 and 20 mg/kg antimony (Figure 4A, 4B). Immunohistochemical analyses demonstrated that GFAP expression in the brain tissue of 20 mg/kg antimony-exposed mice was higher than that in the controls (Figure 4C). Therefore, antimony induces astrocyte activation in vitro and in vivo.
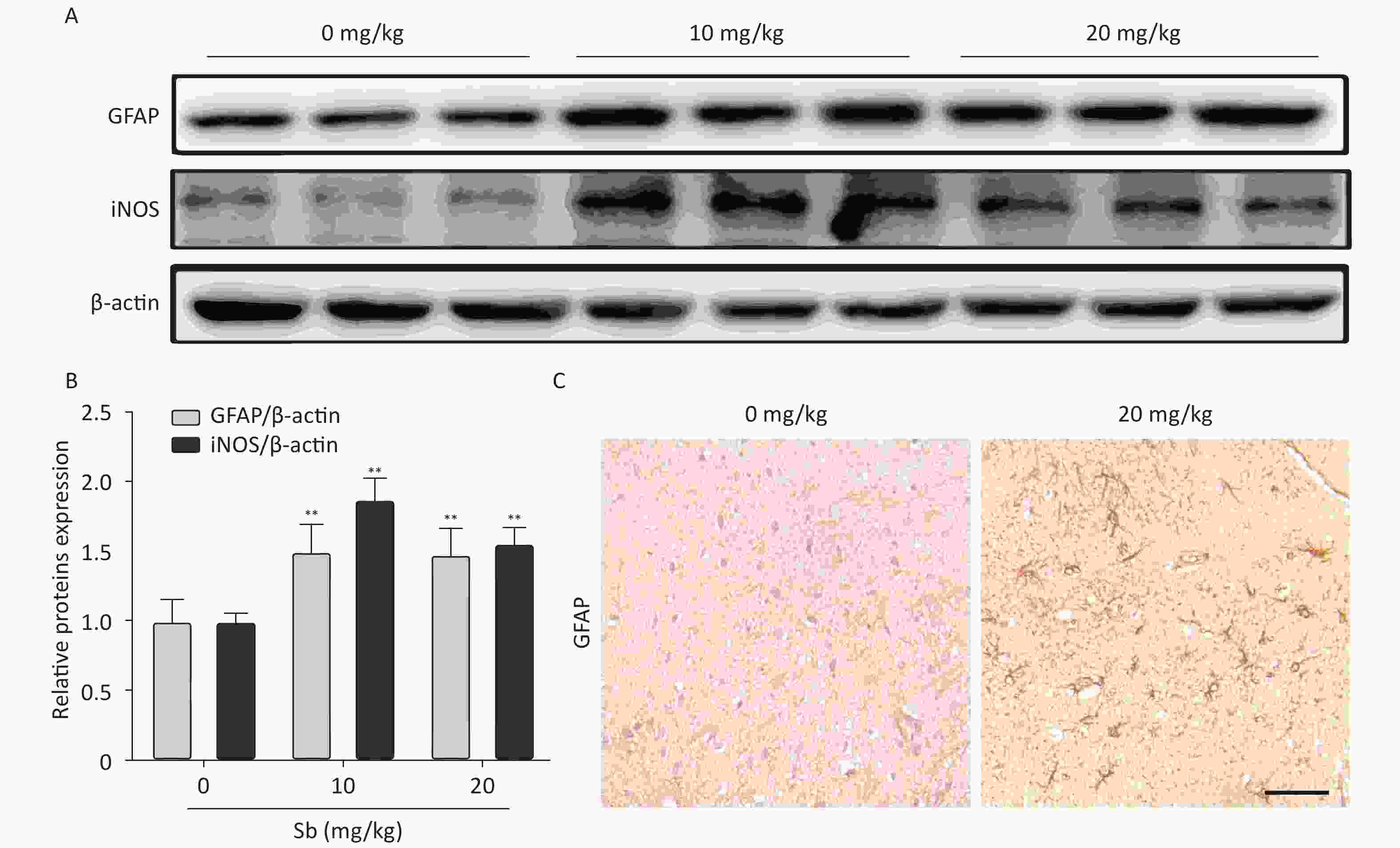
Figure 4. Antimony induces astrocyte activation in murine brains. (A) Mice were treated with different does of antimony for 8 weeks and the levels of GFAP and iNOS proteins in brain tissue were detected by western blot. (B) The levels of GFAP and iNOS proteins were quantified by normalization to β-actin (C) Mice were exposed to antimony at the indicated doses, and the expression of GFAP protein in brain tissue sections was detected by immunohistochemistry. Scale bar: 50 μm. All quantitative data are presented as mean ± SD, **P < 0.01, n = 5.
-
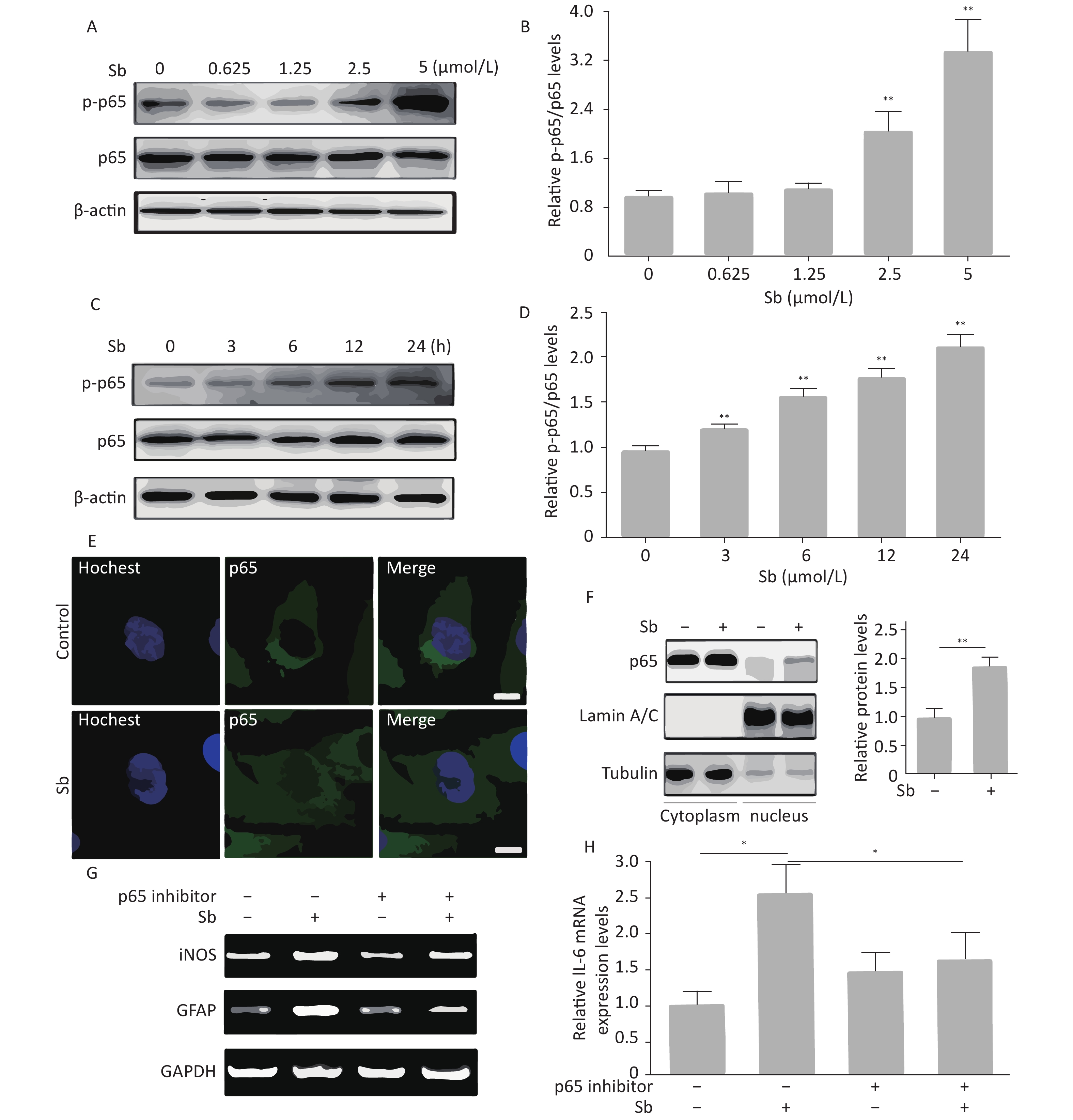
The NF-κB pathway plays a key role in regulating astrocyte activation during various pathological CNS processes[25]. To verify whether the NF-κB pathway is involved in antimony-induced astrocyte activation, we first assessed the effects of antimony on p65 phosphorylation in C6 cells. These experiments demonstrated that p65 phosphorylation was not increased upon exposure to 0.625 or 1.25 μmol/L antimony, while higher doses (2.5 and 5 μmol/L) dramatically stimulated p65 phosphorylation, suggesting antimony-mediated NF-κB pathway activation (Figure 5A, 5B). As shown in Figure 5C, p65 was phosphorylated in a time-dependent manner, and was upregulated within 3 h (Figure 5D). An immunofluorescence assay showed that antimony-induced NF-κB is translocated from the cytoplasm to the nucleus in C6 cells (Figure 5E). This was confirmed by measuring the levels of p65 protein in the nuclear and cytosolic fractions (Figure 5F). Moreover, an NF-κB inhibitor attenuated antimony-mediated upregulation of iNOS and GFAP (Figure 5G). We also found that NF-κB inhibition dramatically decreased IL-6 expression in antimony-exposed C6 cells. Therefore, antimony activates astrocytes via the NF-κB signaling pathway (Figure 5H).
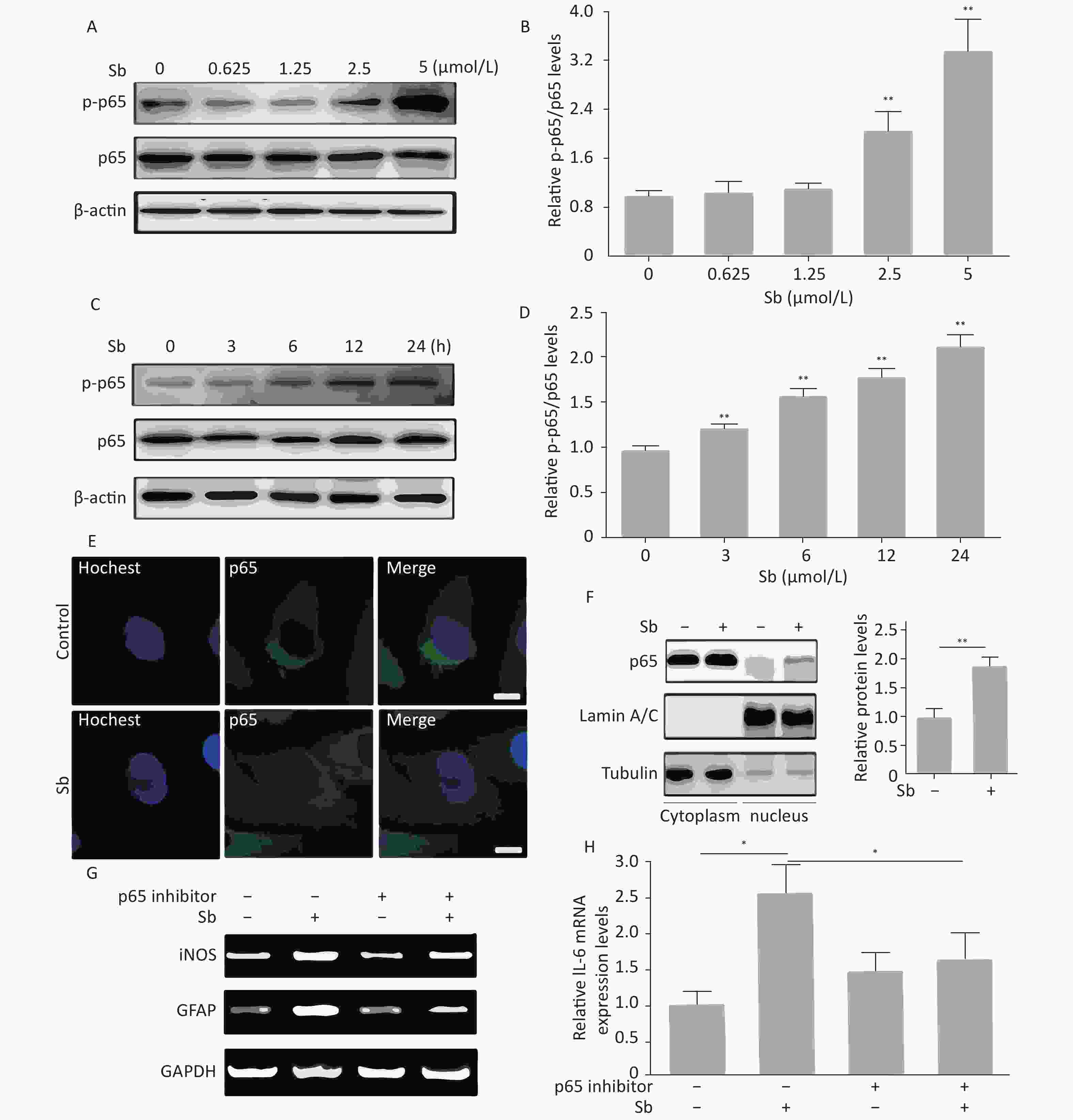
Figure 5. NF-κB signaling is involved in antimony-induced astrocyte activation. (A) C6 cells were treated with antimony at the indicated doses for 24 h, then the levels of cellular NF-κB and p-NF-ΚB proteins were measured by western blot. (B) The levels of NF-κB and p-NF-ΚB proteins in (B) were quantified by normalization to β-actin. (C) C6 cells were treated with antimony (2.5 μmol/L) for varying durations, then the levels of cellular NF-κB and p-NF-κB proteins were measured by western blot. (D) The levels of NF-κB and p-NF-ΚB proteins in (C) were quantified by normalization to β-actin. (E) C6 cells were treated with antimony (2.5 μmol/L) for 24 h, and the localization of cellular p65 was assessed by immunofluorescence. Scale bar: 10 μm. (F) C6 cells were treated with antimony as indicated in (E), then the levels of p65 expression in the cytoplasm and the nucleus were measured by western blot. The graph on the right depicts quantitative analysis of the relative levels of nuclear/cytoplasmic p65 expression. (G, H) C6 cells exposed to antimony were treated with or without pre-treatment with 10 μmol/L BAY11-7082 (NF-κB inhibitor), then the levels of GFAP and iNOS mRNAs were detected by RT-PCR (G). Relative levels of IL-6 mRNA were detected by qRT-PCR (H). Data are presented as mean ± SD, **P < 0.01, *P < 0.05.
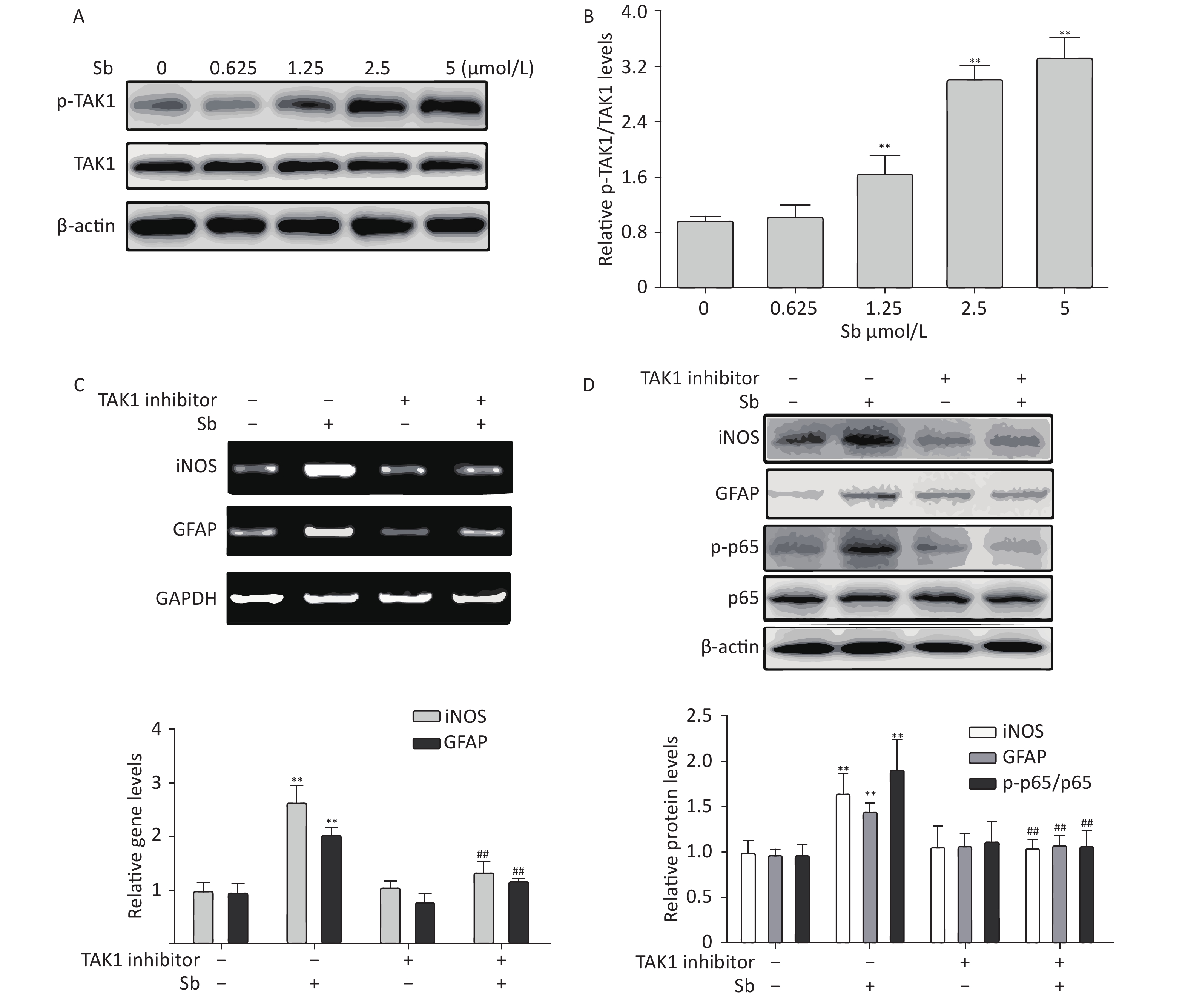
Our group has reported that TAK1 participates in regulating astrocyte activation via the NF-κB pathway[17]. Therefore, we investigated the role of TAK1 in antimony-induced astrocyte activation. To clarify whether antimony exposure stimulates TAK1 phosphorylation, we exposed C6 cells to different doses of antimony and assessed the levels of p-TAK1 and TAK1 proteins. TAK1 phosphorylation was increased by antimony treatment in a dose-dependent manner (Figure 6A, 6B). Moreover, a TAK1 inhibitor alleviated antimony-mediated NF-κB activation and subsequent astrocyte activation, as demonstrated by changes in the expression of iNOS and GFAP proteins and mRNAs (Figure 6C, 6D). Therefore, TAK1 is involved in antimony-mediated astrocyte activation.
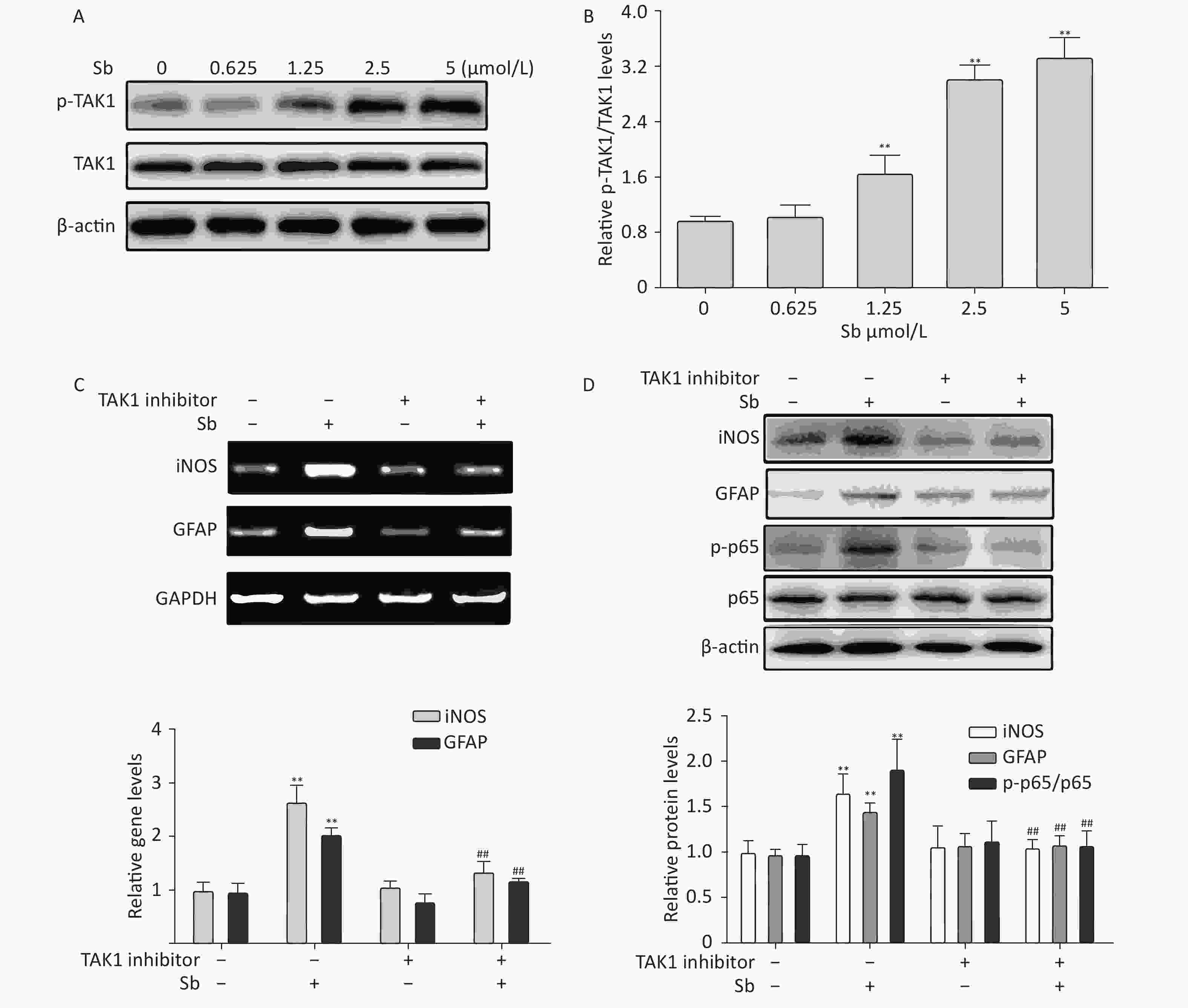
Figure 6. TAK1 is involved in antimony-induced astrocyte activation. (A) C6 cells were treated with antimony at the indicated concentration for 24 h, then the levels of p-TAK1 and TAK1 proteins were measured by western blot. (B) Quantitative analysis of protein expression relative to p-TAK1 in (A). (C) C6 cells exposed to antimony were treated with or without pre-treatment with 20 μmol/L TAK1 inhibitor, then the levels of GFAP and iNOS mRNAs were detected by RT-PCR. The graph below represents statistical analysis of (C). (D) C6 cells were treated with antimony as indicated in (C), then the levels of GFAP, iNOS, p-p65 and p65 proteins were detected by immunoblotting. The graph below represents statistical analysis of (D). Data are presented as mean ± SD, **P < 0.01 vs. control, ##P < 0.01 vs. antimony-exposed group.
-
Studies on antimony in human and mouse brains suggest that antimony is neurotoxic. Oliveira et al.[26] reported that mice exposed to meglumine antimoniate showed changes in several tissues, especially the brain, lung, and heart. Subsequently, Tanu et al. found that antimony led to impaired cognitive function using the Morris water maze. Based on these findings, we found that antimony stimulates neuronal apoptosis, confirming that antimony is a novel nerve poison[4]. Although there is sufficient evidence for antimony-associated neurotoxicity, the results from actual human populations remain limited. One study found higher levels of antimony in the hair of depressed patients[27], suggesting that antimony exposure is related to depression. However, most studies failed to detect differences in antimony concentrations in the plasma, serum, or cerebrospinal fluid of healthy subjects and AD patients[28, 29]. Thus, more evidence is needed, especially in relation to individuals that have experienced occupational exposure. Considering the large number of astrocytes and their biological and pathological effects in the CNS, we examined astrocytes exposed to antimony. A common response to stimulation with environmental toxicants is astrocyte activation, which contributes to CNS damage[30]. The response of astrocytes to CNS damage initiates a process called astrogliosis, which is a pathological hallmark of structural CNS lesions[31]. Enhanced expression of the astrocyte marker GFAP is thought to be a hallmark of astrogliosis, an early neurotoxic event[32]. We found that antimony increased GFAP expression in C6 cells, suggesting that antimony triggers astrocyte activation. In this study, we found that antimony activated astrocytes in a TAK1- and NF-κB-dependent manner. This is the first report of antimony-associated astrocyte activation, providing a new direction for future research on antimony-triggered neurotoxicity.
Different cellular signaling molecules are involved in triggering and regulating reactive astrogliosis, including large polypeptide cytokines, ROS, and Aβ. The responses to different stimuli and intra- and inter-cellular signaling mechanisms involved in the molecular, morphological, and functional changes in reactive astrocytes are context-specific[33]. The main signaling pathways implicated in mediating various aspects of reactive astrogliosis involve signal transducer and activator of transcription 3, NF-κB, and MAPKs. In other cell types, antimony activated c-jun kinase and subsequently triggered apoptosis[34]. Extracellular-signal-regulated kinase, another critical kinase in the MAPK pathway, was activated by antimony to accelerate sirtuin 1 degradation[20]. Therefore, MAPKs might respond to antimony exposure and subsequently activate astrocytes. Interestingly, MAPKs and NF-κB can affect each other and participate in neuroinflammation[35]. Recently, non-coding RNAs, especially circular RNAs, were reported to contribute to astrocyte activation. For example, the circular RNA homeodomain interacting protein kinase 2 (circHIPK2) functions as an endogenous microRNA-124 sponge to sequester microRNA-124 and inhibit its activity, leading to increased expression of sigma non-opioid intracellular receptor 1 and subsequent astrocyte activation[36]. Similarly, circular HECT domain E3 ubiquitin protein ligase 1 functions as an endogenous microRNA-142 sponge to inhibit its activation, contributing to astrocyte activation[37]. Moreover, an increasing number of studies have shown that the gut microbiota plays an important role in communicating between the gastrointestinal tract and the CNS. As the most abundant cells in the CNS, astrocytes are regulated by tryptophan type I interferon and microbial metabolites via aromatic hydrocarbon receptors[38]. The gut microbiota from NLR family pyrin domain containing 3-deficient mice regulates astrocyte dysfunction via circHIPK2[39]. Additional studies are required to identify other signaling pathways and molecules (circular RNA, microRNA, kinases, etc.) involved in antimony-associated cytotoxicity.
Once activated, astrocytes mediate neurotoxicity in several ways. First, astrocyte activation mediates neuroinflammation, an important event in neurodegenerative diseases[40, 41]. Reactive astrocytes produce many molecules with pro- or anti-inflammatory potential, and can have both pro- and anti-inflammatory effects on microglia[42-45]. Moreover, astrocyte activation-mediated inflammatory cytokine upregulation triggers neuronal apoptosis. Conditioned medium of TCDD-exposed astrocytes promoted neuronal apoptosis in a TNF-α-dependent manner[17]. Increased numbers of reactive astrocytes surround amyloid plaques in the AD brain[46]. In reactive astrocytes, the levels of three components involved in Aβ production are increased (amyloid precursor protein, β-secretase, and γ-secretase)[46]. Many animal studies provide experimental evidence that astrocyte activation protects CNS cells through multiple mechanisms, such as protecting cells from oxidative stress via glutathione production[47-49]. Nevertheless, role (s) of antimony-induced astrocyte activation in antimony-triggered neurotoxicity are still unknown and require further investigation.
In summary, we are the first to report that antimony exposure induces astrocyte activation via NF-κB activation. Our data may help to elucidate a novel cellular mechanism underlying antimony-associated neurotoxicity. Our results also shed light on preventive and treatment strategies for antimony neurotoxicity.
doi: 10.3967/bes2021.005
-
Abstract:
Objective Antimony (Sb) has recently been identified as a novel nerve poison, although the cellular and molecular mechanisms underlying its neurotoxicity remain unclear. This study aimed to assess the effects of the nuclear factor kappa B (NF-κB) signaling pathway on antimony-induced astrocyte activation. Methods Protein expression levels were detected by Western blotting. Immunofluorescence, cytoplasmic and nuclear fractions separation were used to assess the distribution of p65. The expression of protein in brain tissue sections was detected by immunohistochemistry. The levels of mRNAs were detected by Quantitative real-time polymerase chain reaction (qRT-PCR) and reverse transcription-polymerase chain reaction (RT-PCR). Results Antimony exposure triggered astrocyte proliferation and increased the expression of two critical protein markers of reactive astrogliosis, inducible nitric oxide synthase (iNOS) and glial fibrillary acidic protein (GFAP), indicating that antimony induced astrocyte activation in vivo and in vitro. Antimony exposure consistently upregulated the expression of inflammatory factors. Moreover, it induced the NF-κB signaling, indicated by increased p65 phosphorylation and translocation to the nucleus. NF-κB inhibition effectively attenuated antimony-induced astrocyte activation. Furthermore, antimony phosphorylated TGF-β-activated kinase 1 (TAK1), while TAK1 inhibition alleviated antimony-induced p65 phosphorylation and subsequent astrocyte activation. Conclusion Antimony activated astrocytes by activating the NF-κB signaling pathway. -
Key words:
- Antimony /
- Astrocyte activation /
- Neurotoxicity /
- NF-κB /
- TAK1
-
Figure 1. Antimony promotes astrocyte proliferation. (A) C6 cells were exposed to antimony at different concentrations (0, 0.01, 0.05, 0.1, 1, 5, 10, 20, 40 μmol/L) for 24 h, and optical density A value of CCK8 regents was used to assess cell proliferation and cytotoxicity. (B) C6 cells were exposed to the indicated antimony concentrations for 24 h, and expression levels of PCNA and cyclin D1 proteins were detected by western blot. (C) The levels of PCNA and cyclin D1 proteins were quantified by normalization to β-actin. Data are presented as mean ± SD, **P < 0.01, *P < 0.05.
Figure 2. Antimony induces astrocyte activation in vitro. (A) C6 cells were exposed to varying concentrations of antimony for 24 h. The expression levels of GFAP and iNOS proteins were measured by immunoblotting. (B) Quantitative analysis of protein expression relative to β-actin expression for (A). (C) C6 cells were exposed to antimony at 2.5 μmol/L, and the proteins expression levels were detected at different times (0, 3, 6, 12, 24 h) by immunoblotting. (D) The levels of GFAP and iNOS proteins were quantified by normalization to β-actin. Data are presented as mean ± SD, **P < 0.01.
Figure 3. Antimony upregulates inflammatory factors in C6 cells. C6 cells were exposed to antimony at the indicated doses, and the levels of IL-1β, IL-6, and TNF-α mRNAs were quantified by qRT-PCR. Data are presented as mean ± SD, **P < 0.01, *P < 0.05.
Figure 4. Antimony induces astrocyte activation in murine brains. (A) Mice were treated with different does of antimony for 8 weeks and the levels of GFAP and iNOS proteins in brain tissue were detected by western blot. (B) The levels of GFAP and iNOS proteins were quantified by normalization to β-actin (C) Mice were exposed to antimony at the indicated doses, and the expression of GFAP protein in brain tissue sections was detected by immunohistochemistry. Scale bar: 50 μm. All quantitative data are presented as mean ± SD, **P < 0.01, n = 5.
Figure 5. NF-κB signaling is involved in antimony-induced astrocyte activation. (A) C6 cells were treated with antimony at the indicated doses for 24 h, then the levels of cellular NF-κB and p-NF-ΚB proteins were measured by western blot. (B) The levels of NF-κB and p-NF-ΚB proteins in (B) were quantified by normalization to β-actin. (C) C6 cells were treated with antimony (2.5 μmol/L) for varying durations, then the levels of cellular NF-κB and p-NF-κB proteins were measured by western blot. (D) The levels of NF-κB and p-NF-ΚB proteins in (C) were quantified by normalization to β-actin. (E) C6 cells were treated with antimony (2.5 μmol/L) for 24 h, and the localization of cellular p65 was assessed by immunofluorescence. Scale bar: 10 μm. (F) C6 cells were treated with antimony as indicated in (E), then the levels of p65 expression in the cytoplasm and the nucleus were measured by western blot. The graph on the right depicts quantitative analysis of the relative levels of nuclear/cytoplasmic p65 expression. (G, H) C6 cells exposed to antimony were treated with or without pre-treatment with 10 μmol/L BAY11-7082 (NF-κB inhibitor), then the levels of GFAP and iNOS mRNAs were detected by RT-PCR (G). Relative levels of IL-6 mRNA were detected by qRT-PCR (H). Data are presented as mean ± SD, **P < 0.01, *P < 0.05.
Figure 6. TAK1 is involved in antimony-induced astrocyte activation. (A) C6 cells were treated with antimony at the indicated concentration for 24 h, then the levels of p-TAK1 and TAK1 proteins were measured by western blot. (B) Quantitative analysis of protein expression relative to p-TAK1 in (A). (C) C6 cells exposed to antimony were treated with or without pre-treatment with 20 μmol/L TAK1 inhibitor, then the levels of GFAP and iNOS mRNAs were detected by RT-PCR. The graph below represents statistical analysis of (C). (D) C6 cells were treated with antimony as indicated in (C), then the levels of GFAP, iNOS, p-p65 and p65 proteins were detected by immunoblotting. The graph below represents statistical analysis of (D). Data are presented as mean ± SD, **P < 0.01 vs. control, ##P < 0.01 vs. antimony-exposed group.
Table 1. Oligonucleotide sequences used in PCR
Gene name Primer name Sequence (5’–3’) RAT-GAPDH GAPDH-F TGATGACATCAAGAAGGTGGTGAAG GAPDH-R TCCTTGGAGGCCATGTGGGCCAT RAT-iNOS iNOS-F GCTTTGTGCGGAGTGTCAGT iNOS-R CCTCCTTTGAGCCCTCTGTG RAT-GFAP GFAP-F GGCGCTCAATGCTGGCTTCA GFAP-R TCTGCCTCCAGCCTCAGGTT RAT-IL-6 IL-6-F GAAAGTCAACTCCATCTGCC IL-6-R CATAGCACACTAGGTTTGCC RAT- IL-1β IL-1β-F TCAGGAAGGCAGTGTCACTCATTG IL-1β-R ACACACTAGCAGGTCGTCATCATC RAT-TNF-α TNF-α-F GCCGATTTGCCATTTCAT TNF-α-R CAGTCGCCTCACAGAGCAA  下载: 导出CSV
下载: 导出CSV
-
[1] Sundar S, Chakravarty J. Antimony toxicity. Int J Environ Res Public Health, 2010; 7, 4267−77. [2] Höck A, Demmel U, Schicha H, et al. Trace element concentration in human brain: activation analysis of cobalt, iron, rubidium, selenium, zinc, chromium, silver, cesium, antimony and scandium. Brain, 1975; 98, 49−64. [3] Tanu T, Anjum A, Jahan M, et al. Antimony-induced neurobehavioral and biochemical perturbations in mice. Biol Trace Elem Res, 2018; 186, 199−207. [4] Wang XK, Zhu PY, Xu SY, et al. Antimony, a novel nerve poison, triggers neuronal autophagic death via reactive oxygen species-mediated inhibition of the protein kinase B/mammalian target of rapamycin pathway. Int J Biochem Cell Biol, 2019; 114, 105561. [5] Mao JM, Yang JB, Zhang Y, et al. Arsenic trioxide mediates HAPI microglia inflammatory response and subsequent neuron apoptosis through p38/JNK MAPK/STAT3 pathway. Toxicol Appl Pharmacol, 2016; 303, 79−89. [6] Phuagkhaopong S, Ospondpant D, Kasemsuk T, et al. Cadmium-induced IL-6 and IL-8 expression and release from astrocytes are mediated by MAPK and NF-κB pathways. Neurotoxicology, 2017; 60, 82−91. [7] Maiuolo J, Macrì R, Bava I, et al. Myelin disturbances produced by sub-toxic concentration of heavy metals: the role of oligodendrocyte dysfunction. Int J Mol Sci, 2019; 20, 4554. [8] Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol, 2015; 7, a020412. [9] Blanco-Suárez E, Caldwell ALM, Allen NJ. Role of astrocyte-synapse interactions in CNS disorders. J Physiol, 2017; 595, 1903−16. [10] Dossi E, Vasile F, Rouach N. Human astrocytes in the diseased brain. Brain Res Bull, 2018; 136, 139−56. [11] Carter SF, Herholz K, Rosa-Neto P, et al. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol Med, 2019; 25, 77−95. [12] Jiao M, Yin KZ, Zhang T, et al. Effect of the SSeCKS-TRAF6 interaction on gastrodin-mediated protection against 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced astrocyte activation and neuronal death. Chemosphere, 2019; 226, 678−86. [13] Chen XX, Nie XK, Mao JM, et al. Perfluorooctane sulfonate mediates secretion of IL-1β through PI3K/AKT NF-кB pathway in astrocytes. Neurotoxicol Teratol, 2018; 67, 65−75. [14] Guénette SY. Astrocytes: a cellular player in Aβ clearance and degradation. Trends Mol Med, 2003; 9, 279−80. [15] Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev, 2017; 277, 113−27. [16] Dresselhaus EC, Meffert MK. Cellular specificity of NF-κB function in the nervous system. Front Immunol, 2019; 10, 1043. [17] Wan CH, Zhang Y, Jiang JK, et al. Critical role of TAK1-dependent nuclear factor-κB signaling in 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced astrocyte activation and subsequent neuronal death. Neurochem Res, 2015; 40, 1220−31. [18] Liu XH, Buffington JA, Tjalkens RB. NF-κB-dependent production of nitric oxide by astrocytes mediates apoptosis in differentiated PC12 neurons following exposure to manganese and cytokines. Mol Brain Res, 2005; 141, 39−47. [19] Dieter MP, Jameson CW, Elwell MR, et al. Comparative toxicity and tissue distribution of antimony potassium tartrate in rats and mice dosed by drinking water or intraperitoneal injection. J Toxicol Environ Health, 1991; 34, 51−82. [20] Zhao XY, Jin Y, Yang LJ, et al. Promotion of SIRT1 protein degradation and lower SIRT1 gene expression via reactive oxygen species is involved in Sb-induced apoptosis in BEAS-2b cells. Toxicol Lett, 2018; 296, 73−81. [21] Zhao XY, Wu YF, Li JL, et al. JNK activation-mediated nuclear SIRT1 protein suppression contributes to silica nanoparticle-induced pulmonary damage via p53 acetylation and cytoplasmic localisation. Toxicology, 2019; 423, 42−53. [22] Piccolini VM, Esposito A, Dal Bo V, et al. Cerebellum neurotransmission during postnatal development: [Pt(O,O'-acac)(γ-acac)(DMS)] vs. cisplatin and neurotoxicity. Int J Dev Neurosci, 2015; 40, 24−34. [23] Baldin V, Lukas J, Marcote MJ, et al. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev, 1993; 7, 812−21. [24] Dietrich DR. Toxicological and pathological applications of proliferating cell nuclear antigen (PCNA), a novel endogenous marker for cell proliferation. Crit Rev Toxicol, 1993; 23, 77−109. [25] Olabarria M, Goldman JE. Disorders of astrocytes: alexander disease as a model. Annu Rev Pathol, 2017; 12, 131−52. [26] Oliveira LFG, Souza-Silva F, Cysne-Finkelstein L, et al. Evidence for Tissue Toxicity in BALB/c Exposed to a Long-Term Treatment with Oxiranes Compared to Meglumine Antimoniate. Biomed Res Int, 2017; 2017, 9840210. [27] Saghazadeh A, Rezaei N. Systematic review and meta-analysis links autism and toxic metals and highlights the impact of country development status: higher blood and erythrocyte levels for mercury and lead, and higher hair antimony, cadmium, lead, and mercury. Prog Neuro-Psychopharmacol Biol Psychiatry, 2017; 79, 340−68. [28] Gerhardsson L, Lundh T, Minthon L, et al. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer's disease. Dement Geriatr Cogn Disord, 2008; 25, 508−15. [29] Alimonti A, Ristori G, Giubilei F, et al. Serum chemical elements and oxidative status in Alzheimer's disease, Parkinson disease and multiple sclerosis. NeuroToxicology, 2007; 28, 450−6. [30] Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol, 2016; 275, 305−15. [31] Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol, 2014; 7, a020420. [32] Brenner M. Role of GFAP in CNS injuries. Neurosci Lett, 2014; 565, 7−13. [33] Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci, 2009; 32, 638−47. [34] Mann KK, Davison K, Colombo M, et al. Antimony trioxide-induced apoptosis is dependent on SEK1/JNK signaling. Toxicol Lett, 2006; 160, 158−70. [35] Ge X, Zhang DM, Li MM, et al. Microglial LOX-1/MAPKs/NF-κB positive loop promotes the vicious cycle of neuroinflammation and neural injury. Int Immunopharmacol, 2019; 70, 187−200. [36] Huang RR, Zhang Y, Han B, et al. Circular RNA HIPK2 regulates astrocyte activation via cooperation of autophagy and ER stress by targeting MIR124-2HG. Autophagy, 2017; 13, 1722−41. [37] Han B, Zhang Y, Zhang YH, et al. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142-TIPARP: implications for cerebral ischemic stroke. Autophagy, 2018; 14, 1164−84. [38] Rothhammer V, Mascanfroni ID, Bunse L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med, 2016; 22, 586−97. [39] Zhang Y, Huang RR, Cheng MJ, et al. Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome, 2019; 7, 116. [40] Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol, 2016; 37, 608−20. [41] Chen WW, Zhang X, Huang WJ. Role of neuroinflammation in neurodegenerative diseases (Review). Mol Med Rep, 2016; 13, 3391−96. [42] Eddleston M, Mucke L. Molecular profile of reactive astrocytes-implications for their role in neurologic disease. Neuroscience, 1993; 54, 15−36. [43] John GR, Lee SC, Brosnan CF. Cytokines: powerful regulators of glial cell activation. Neuroscientist, 2003; 9, 10−22. [44] Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol, 2007; 28, 138−45. [45] Min KJ, Yang MS, Kim SU, et al. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci, 2006; 26, 1880−7. [46] Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol, 2017; 7, 170228. [47] Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol, 2010; 119, 7−35. [48] Vargas MR, Johnson DA, Sirkis DW, et al. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci, 2008; 28, 13574−81. [49] Chen YM, Vartiainen NE, Ying WH, et al. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J Neurochem, 2001; 77, 1601−10. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2421
- HTML全文浏览量: 1069
- PDF下载量: 107
- 被引次数: 0

 Quick Links
Quick Links