

 下载:
下载:

-
Dioxins are one of the most poisonous organic pollutants of natural and anthropogenic origin that are persistent in the environment. The compound 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) is ubiquitous in the environment and causes detrimental health effects through bioaccumulation in humans via the food chain[1]. After being internalized into the body, it mainly targets the central nervous system (CNS) organs. A recent study reported the TCDD-mediated impairment in neurocognitive functions after 50 years from exposure[2]. Furthermore, different types of CNS cells respond to TCDD exposure, including neurons, microglial cells, and astrocytes[3-5].
Previously we have reported the effects of TCDD on astrocytes, the most abundant glial cell type in the brain. Astrocytes have been traditionally defined as the passive housekeepers that preserve the optimal microenvironment and ensure normal neuronal functioning. Astrocyte dysfunction is involved in various CNS disorders such as Alzheimer's disease[6], amyotrophic lateral sclerosis[7], cerebral ischemia[8]. During these pathological processes, astrocytes undergo astrogliosis, a complex response comprising cellular hypertrophy, proliferation, and dysfunction[9]. Reactive astrocytes are likely to release a variety of molecules such as interleukin (IL)-6, IL-1β, tumor necrosis factor (TNF)-α, and nitric oxide (NO), which affect the functioning of the surrounding cells[10]. We have shown that TCDD exposure promotes the activation of astrocytes and enhances the secretion of TNF-α and NO via the protein kinase C (PKC)/Src-suppressed C-kinase substrate (SSeCKS)-dependent mechanism[11]. We have also reported that the transforming growth factor beta-activated kinase 1 (TAK1)-dependent nuclear factor kappa B (NF-κB) signaling participates in the TCDD-induced astrocyte activation[3].
Cell proliferation, a marker of astrogliosis, was reported to be involved in the TCDD-associated toxicity. TCDD exhibited pro-proliferation effects, such as evident from the stimulation of the proliferation of HAPI microglia, through its effect on the glycogen synthase kinase (GSK)-3β signaling pathway[5]. Furthermore, TCDD also suppressed the growth of HepG2 cells in vitro[12]. Li et al. reported the TCDD-mediated inhibition of human ovarian cancer cell proliferation via the aryl hydrocarbon receptor (AHR) signaling pathway[13]. The proliferation of SK-N-SH human neuronal cells was also inhibited by TCDD exposure[14]. TCDD, as a multiple-site and multiple-species carcinogen, could induce cancer in multiple organs. Cancer progression involves multiple mechanisms, including apoptosis inhibition, cell proliferation, invasion, and angiogenesis. Therefore, the investigation of the effects of TCDD on the proliferation of astrocytes may improve our understanding of the TCDD-associated astrogliosis and carcinogenesis. Various studies have suggested that the signal transducer and activator of transcription 3 (STAT3) is an important signaling molecule in astrogliosis[15, 16]. This protein plays an important role in cellular proliferation, a critical process in astrogliosis. A recent report suggested that STAT3 directly binds to the promoter of cyclin D1, a member of the cell cycle regulating protein, and promotes its transcription[17, 18]. The serine/threonine kinase, Akt, also known as protein kinase B (PKB), plays key roles in cell survival, growth, and proliferation[19]. It promotes cell cycle progression, mainly by inducing the cell cycle transition from the G1 to S phase. Furthermore, Akt acts upstream of STAT3, and may activate STAT3[20]. Akt activation was observed in different cells in response to TCDD exposure[5, 21, 22].
In the present study, we investigated the effects of TCDD on the Akt/STAT3 pathway in astrocytes and revealed the involvement of this pathwayin TCDD-induced cell proliferation. TCDD induced the proliferation of astrocytes via the Akt/STAT3/cyclin D1 pathway activation. Our results provide a novel insight into the mechanism underlying the TCDD-induced neurotoxicity.
-
The regents were purchased from following commercial suppliers: TCDD (Sigma, F-402S), LY294002 (Selleck, S1105), and AG490 (Beyotime, S1509).
The antibodies used were as follows: anti-p-Akt (Cell Signaling, 4060), anti-Akt (Cell Signaling, 4691), anti-p-STAT3 (Cell Signaling, 9145), anti-STAT3 (Cell Signaling, 9139), anti-CyclinD1 (Cell Signaling, 2978), anti-GAPDH (Sigma, G9545), anti-Histon H3 (Cell Signaling, 14269), anti-Tubulin (proteintech, 10068-1-AP).
-
Primary astrocytes were isolated from the cerebral cortex of newborn Sprague-Dawley rats at postnatal day 0-1 (supplied by the University Laboratory Animal Services Centre, The Nantong University), as previously described[11]. All animal procedures were approved by the Animal Experimental Committee of Nantong University. Briefly, the brain tissue was harvested and dissected in ice-cold phosphate-buffered saline (PBS). Meninges were carefully removed and the neopallium was isolated. The tissues were cut into 1 mm cubes and digested with 0.25% trypsin for 15 min at 37 ℃. The cells were passed through a nylon mesh of 70-mm pore size. The isolated cells were cultured in 75 cm2 Falcon culture flasks at a density of 2 × 107 cells/flask. These cells were cultured for 7-10 days until confluency following the protocol used for C6 cell cultivation. To obtain a more homogeneous layer of astrocytes, the flasks were placed on a horizontal shaker and shaken for 18 h at 180 rpm. The shaker temperature was maintained at 37 ℃. After 8 h, the supernatant containing loose astrocytes and oligodendrocytes was discarded. The purity of the astrocytes was found to be 95%, as verified with immunocytochemical staining using glial fibrillary acidic protein (GFAP) monoclonal antibody (data not shown).
Rat C6 astrocyte cells were purchased from the American Type Culture Collection and cultured as previously described[23]. Briefly, the cells were cultured in Dulbecco's modified Eagle's medium (Gibco, C11965500BT) supplemented with 10% fetal bovine serum (Gibco, 1027-106) at 37 ℃ in an humidified atmosphere of 95% air and 5% CO2. The C6 cells were plated in 24-well plates for 24 h before TCDD treatment, followed by the exposure to TCDD at concentrations ranging from 0.1 to 100 nmol/L[11, 23]. The cells were pretreated for 1 h with LY294002 (50 μmol/L) or AG490 (50 µmol/L) to inhibit the activity of Akt or STAT3, respectively.
-
The EdU assay was used to assess cell proliferation with Cell-LightTM EdU DNA Cell Proliferation Kit (RiboBio, C10310) according to the manufacturer's protocol. In brief, the C6 cells were plated in 24-well plates for 24 h before TCDD treatment. After TCDD treatment, 50 μmol/L EdU was added to the cells for another 2 h. The cells were fixed with 4% formaldehyde for 30 min, and labeled with Apollo® for 30 min and Hoechst 33342 for 10 min. The cells were visualized with a microscope (Leica, DM4000B).
-
Propidium iodide (PI) staining was used to evaluate cell cycle stages. After treatment, the cells were harvested and resuspended in pre-cooled 70% ethanol. The cells were stored at -20 ℃ overnight, followed by incubation with 50 μg/mL PI (Becton-Dickinson, San Jose, CA) in PBS containing 1 mg/mL RNase A at 25 ℃ for 30 min. The cell cycle distribution was analyzed using a Becton-Dickinson BD Fascine flow cytometry and Cell Quest acquisition and analysis software (Becton Dickinson, Franklin Lakes, NJ).
-
Total cellular proteins were collected and subjected to immunoblotting as previously described[24]. For Western blot analysis, aliquots of 20 μg of protein were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane filters. The membranes were incubated with 3% bovine serum albumin. Several primary antibodies (GAPDH, 1:2000; Akt, 1:1000; p-Akt, 1:1000; STAT3, 1:1000; p-STAT3, 1:1000; cyclin D1, 1:1000) were added to the blot at 4 ℃ overnight, followed by treatment with corresponding secondary antibodies for another 1 h at room temperature. The relative protein expression was quantified with densitometry after visualization (ImageJ, NIH, Bethesda, MD).
-
After treatment, the C6 cells on coverslips were fixed with 4% paraformaldehyde for 15 min and washed thrice with PBS. The cells were permeabilized with 0.2% Triton X-100 for another 15 min at 4 ℃. The cells were blocked with goat serum and incubated with anti-STAT3 (1:200) at 4 ℃ overnight, followed by treatment with Hoechst for the visualization of nuclei. The glass slide was overlaid with a coverslip using glycerol and the cells were visualized under a fluorescence microscope (Leica, DM4000B).
-
After cell collection, we performed cytoplasmic and nuclear fractions separation by using nuclear and cytoplasmic protein extraction kit (Beyotime, P0027). In brief, cells treated as indication were resuspended in PBS, followed by addition to cytoplasmic protein extraction reagent A and B. Then, the samples were centrifuged, and the supernatant was collected to obtain cytoplasmic fraction. The nuclei was resuspended in nuclear protein extraction agent. After centrifugation, the supernatant was collected as the nuclear fraction.
-
Data are expressed as mean ± standard error of the mean (SEM) for at least three independent experiments. Differences in each group were evaluated with one-way analysis of variance (ANOVA). All data were analyzed with Stata 17.0 statistical software (Stata Corp, College Station, TX). A value of P ≤ 0.05 was considered significant.
-
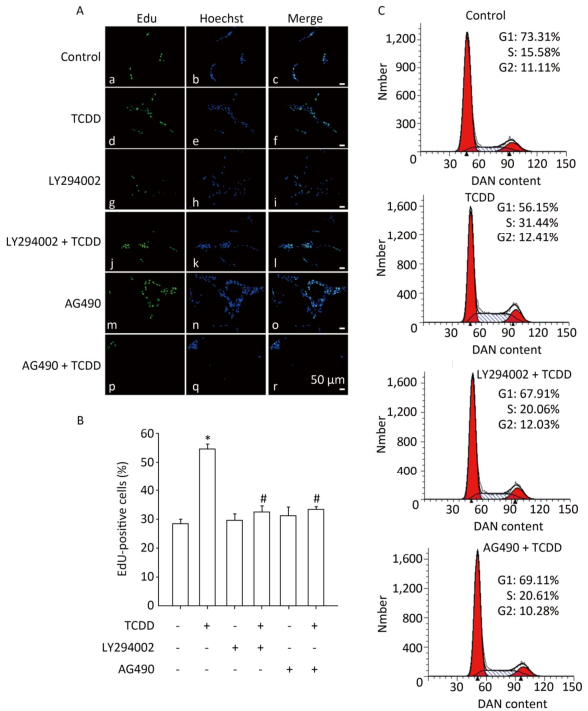
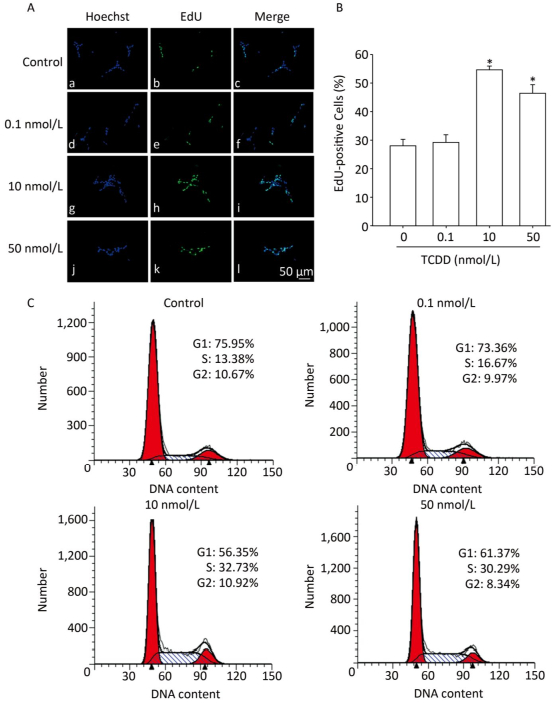
To assess the effects of TCDD on the proliferation of C6 cells, we performed the EdU-based proliferation assay after TCDD treatment at different concentrations. The percentage of EdU-positive cells was significantly higher in the group treated with 10 and and 50 nmol/L of TCDD for 24 h than in the control group (Figure 1A and B). However, low dose (0.1 nmol/L) of TCDD had no effect on the percentage of EdU-positive cells. To determine the percentage of cells in different phases of cell cycle, PI staining was carried out and the cells were analyzed with flow cytometry. After exposure to 10 or 50 nmol/L TCDD for 24 h, the number of cells in the G1 phase decreased and that of cells in the S phase increased (Figure 1C). Taken together, these data show that TCDD stimulates the proliferation of C6 cells.
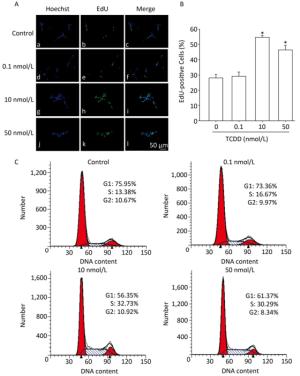
Figure 1. TCDD induces proliferation of C6 cells. (A) C6 cells were treated with DMSO or 0.1, 10, and 50 nmol/L TCDD for 24 h and subjected to EdU-based proliferation assay. (B) Quantitative analysis of the percentage of EdU-positive cells after TCDD treatment. (C) Flow cytometry analysis was performed to examine the cell cycle phase of C6 glioma cells after treatment with 0, 0.1, 10, and 50 nmol/L TCDD for 24 h. Scale bars: 50 μm. (*P < 0.05, significantly different from the control group).
-
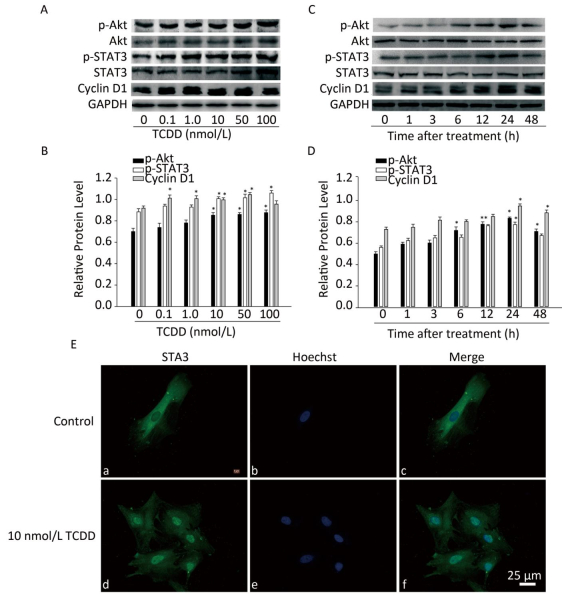
Next, we characterized the molecular mechanisms underlying the increase in the proliferation of C6 cells induced by TCDD. As cyclin D1 is an important protein regulating cell cycle and is involved in the G1/S transition, we first measured cyclin D1 expression level using western blotting and found that TCDD exposure resulted in an increase in cyclin D1 levels in a dose- and time-dependent manner (Figure 2). STAT3 participates in astrogliosis by binding to the cyclin D1 promoter and promotes its transcription[17]. Akt also promotes cell cycle progression mainly through its involvement in the transition of the cells from G1 to S phase and functions upstream of cyclin D1. Therefore, we examined the activation of Akt and STAT3 after treating cells with TCDD. As shown in Figure 2A and B, TCDD exposure induced a significant increase in the levels of p-Akt and p-STAT3 in a dose-dependent manner. We also measured the time-dependent effects of Akt and STAT3 activation and found that p-Akt level increased as early as 6 h, while p-STAT3 expression increased after 6 h. Thus, STAT3 activation occurred after Akt expression induction (Figure 2C and D). We also measured the distribution of STAT3 in primary astrocytes. In the control group, STAT3 was mainly distributed in the cytoplasm, but it rapidly translocated from the cytoplasm to the nucleus after TCDD treatment. This observation confirms the activation of STAT3 after the treatment of cells with 10 nmol/L TCDD (Figure 2E).
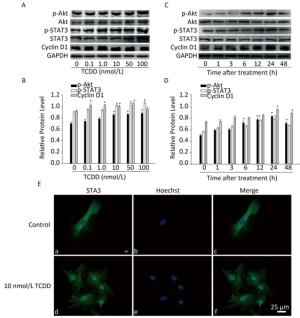
Figure 2. TCDD activates Akt and STAT3 and increases cyclin D1 expression. (A) The protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH in C6 cells after exposure to different doses of TCDD for 24 h. (B) Quantitative analysis of protein expression relative to GAPDH expression for (A). (C) The protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH in C6 cells after exposure to 10 nmol/L TCDD for indicated time periods. (D) Quantitative analysis of protein expression relative to GAPDH expression for (C). (E) Immunofluorescence staining of STAT3. Primary astrocytes were treated with either 0.1% DMSO (a-c; controls) or 10 nmol/L TCDD (d-f) for 24 h. Nucleus was stained with Hoechst (blue fluorescence); STAT3 was stained with green fluorescence. Scale bars: 25 μm. (*P < 0.05, significantly different from the DMSO-treated group).
-
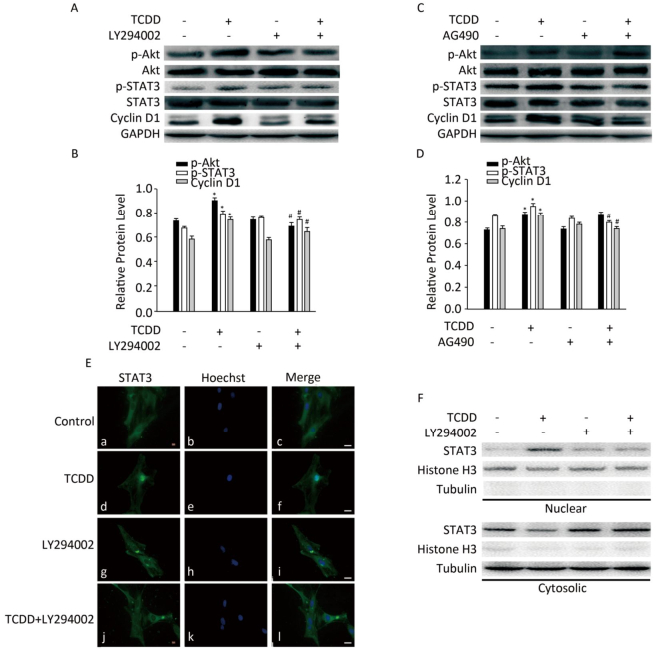
To investigate the involvement of the Akt/STAT3 pathway in cyclin D1 upregulation after TCDD treatment, we treated cells with different inhibitors prior to TCDD stimulation. First, we used LY294002 to inhibit the activity of Akt, and measured the levels of p-Akt, p-STAT3, and cyclin D1. As a result, we observed elevated expression levels of p-Akt and p-STAT3 and attenuation of cyclin D1 expression (Figure 3A and B). AG490, a JAK2 inhibitor, was used to inhibit the activation of STAT3. However, AG490 only downregulated the expression of p-STAT3 and cyclin D1 in TCDD-treated cells, while the level of p-Akt was unaffected. These results suggest that Akt is the upstream regulator of the TCDD-induced phosphorylation of STAT3 (Figure 3C and D). The nuclear distribution of STAT3 significantly decreased after the treatment of primary astrocytes with LY294002 (Figure 3E). We also observed STAT3 nuclear translocation after the treatment of the cells with TCDD, and this phenomenon was dramatically inhibited by LY294002. Taken together, these results indicate that the TCDD-mediated phosphorylation of Akt contributes to STAT3 activation, eventually leading an increase in the expression of cyclin D1.
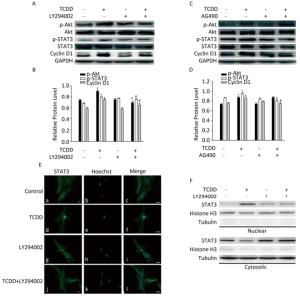
Figure 3. Akt/STAT3/cyclin D1 pathway is activated after TCDD treatment. (A) C6 cells were pre-treated with Akt inhibitor LY294002 (50 μmol/L) for 1 h prior to TCDD stimulation. Protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH were analyzed with western blotting. (B) Quantitative analysis of protein expression relative to GAPDH expression for (A). (C) C6 cells were pre-treated with STAT3 inhibitor AG490 (50 μmol/L) for 1 h prior to TCDD stimulation. Protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH were analyzed with western blotting. (D) Quantitative analysis of protein expression relative to GAPDH expression for (C). (E) Primary astrocytes were treated with 0.1% DMSO (a-c; controls), 10 nmol/L TCDD (d-f), LY294002 (g-i), and LY294002 and TCDD (j-l) for 24 h. Nucleus was stained with Hoechst (blue fluorescence), and STAT3 was stained with green fluorescence. (F) C6 cells were treated with TCDD as indicated in (E), and STAT3 expression in the nucleus or cytoplasm was detected with western blotting. Scale bars: 25 μm. (*P < 0.05, significantly different from the control; #P < 0.05, significantly different from TCDD only group).
-
To investigate the contribution of the Akt/STAT3 signaling pathway in the TCDD-induced astrocyte proliferation, EdU-based proliferation assay and PI staining analysis with flow cytometry were conducted after the pretreatment of cells with LY294002 or AG490. Both LY294002 and AG490 reduced the TCDD-mediated increase in C6 cell proliferation. Furthermore, the TCDD-induced transition of cells from G0/G1 to S phase was effectively prevented in the presence of the two inhibitors (Figure 4).
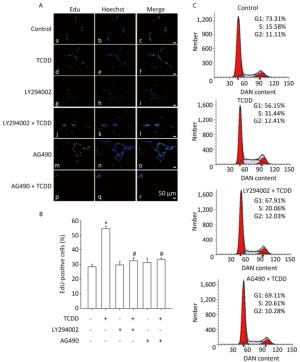
Figure 4. The Akt/STAT3 pathway is involved in the TCDD-mediated proliferation of C6 cells. (A) EdU-based proliferation assay was performed to estimate the proliferation rate of C6 cells. Cells were pre-treated with an Akt inhibitor LY294002 (50 μmol/L) or a STAT3 inhibitor AG490 (50 μmol/L) for 1 h prior to TCDD stimulation. (B) Quantitative analysis of EdU-positive cells after TCDD treatment. (C) Flow cytometry analysis showed the cell cycle phase of C6 cells after treatment with 0.1% DMSO, 10 nmol/L TCDD, LY294002 and TCDD, and AG490 and TCDD for 24 h. Scale bars: 50 μm. (*P < 0.05, significantly different from the control group; #P < 0.05, significantly different from TCDD only group).
-
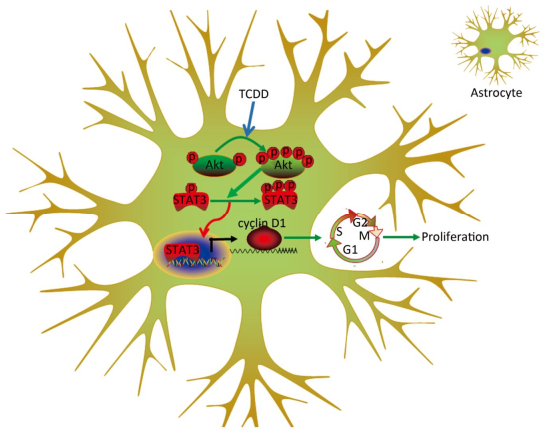
Animal experiments and population-based human studies have shown that TCDD exposure leads to neurological disorders. Several studies have been directed to understand the mechanism underlying the neurotoxicity induced by TCDD. In the present study, we assessed the effects of TCDD on astrocyte proliferation and revealed the underlying molecular mechanism of action. As a result, we found that TCDD triggered astrocyte proliferation and increased the expression of cyclin D1 through the activation of the Akt/STAT3 pathway. These effects were prevented by the inhibition of the Akt signaling. Our findings provide the first evidence of the effects and underlying mechanism of action of TCDD on astrocyte proliferation (Figure 5).
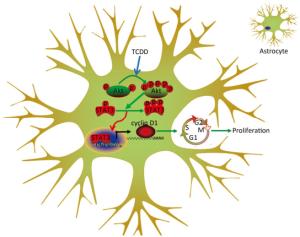
Figure 5. Schematic diagram deciphering the mechanism underlying the Akt/STAT3-mediated cell proliferation by TCDD in astrocyte cells.
Considering the importance of proliferation in astrogliosis and carcinogenesis, we assessed the effects of TCDD on the proliferation of astrocytes and found that TCDD accelerated astrocyte proliferation via the Akt/STAT3/cyclin D1 pathway. Recent evidence suggested the important role of STAT3 in proliferation and astrogliosis[25-27]. At the molecular level, STAT3 directly binds to the promoter of cyclin D1 and induces its transcription[17]. STAT3 is also a key signaling molecule involved in mediating the toxicity induced by TCDD[28, 29]. Akt regulates cell survival, growth, and proliferation via STAT3 activation. Li et al. reported that Akt serves as an upstream regulator of STAT3 and mediates B7-H3-promoted cellular migration[30]. However, STAT3 may also act as an upstream regulator of Akt[31]. We determined the expression levels of p-Akt, and found that the TCDD-induced elevation in p-Akt levels occurred in a dose- and time-dependent manner (Figure 2). Moreover, our results showed that Akt was an upstream regulator of STAT3 because Akt inhibition completely abolished the TCDD-mediated increase in p-STAT3 expression. On the contrary, AG490 had no effect on the expression of p-Akt (Figure 3). Furthermore, LY294002 attenuated the TCDD-mediated nuclear translocation of STAT3 (Figure 3).
NF-κB, a classic inflammatory regulator, was activated in response to TCDD treatment and was involved in astrocyte activation[3, 11]. NF-κB activation is also implicated in the acceleration of cell proliferation and is mediated via TNF-α and IL-6[32]. Guttridge et al. identified cyclin D1 as an important transcriptional target of NF-κB, and revealed the mechanism that explains the involvement of NF-κB in cell growth regulation[33]. Several reports have provided evidence on the effect of TCDD on reactive oxygen species (ROS) generation. TCDD exposure increases ROS production in PC12 cells, astrocytes, and SH-SY5Y cells[34-36]. An increase in the level of ROS production has also been detected in various cancers through the activation of multiple pro-tumorigenic processes, including enhanced cell survival and proliferation[37]. ROS plays a central role in palmitic acid-stimulated hepatocyte proliferation through Akt activation[38] and contributes to STAT3 activation under specific conditions[39]. Taken together, we suggest that other mechanisms, including NF-κB activation and ROS production, may possibly contribute to the TCDD-mediated astrocyte proliferation.
Several factors may affect the toxicity of TCDD. First, the effects of TCDD on cell proliferation may vary in different research models. As mentioned before, the proliferation of different cell types was affected by TCDD treatment. The second influential factor is the exposure condition. Reale et al. showed that the effects of fetal exposure of TCDD on the thyroid function varied with different doses of TCDD[40]. Finally, the metabolism is also potential factor involved in TCDD-associated toxicity. Sorg et al. identified and measured TCDD metabolites in a patient with TCDD poisoning, and found that most TCDD (60%) was not modified after elimination. Two TCDD metabolites were identified in the feces, blood serum, and urine[41]. Whether TCDD metabolites differ in various organs is, however, questionable.
In summary, TCDD induces the proliferation of astrocytes through the Akt/STAT3/cyclin D1 pathway. As proliferation is an important characteristic of astrogliosis, our results provide a strong evidence of a novel mechanism underlying the TCDD-mediated neurotoxicity. Further in vivo studies are warranted to confirm our findings.
No potential conflicts of interest were disclosed.
doi: 10.3967/bes2019.038
2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin Promotes Proliferation of Astrocyte Cells via the Akt/STAT3/Cyclin D1 Pathway
-
Abstract:
Objective The compound 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD), a persistent organic pollutant, is harmful to the nervous system, but its effects on the brain are still unclear. This study aimed to investigate the effects of TCDD on astrocytes proliferation and underlying molecular mechanism. Methods The cell proliferation was measured by EdU-based proliferation assay and PI staining by flow cytometry. Protein expression levels were detected by Western blotting. Immunofluorescence, cytoplasmic and nuclear fractions separation were used to assess the distribution of signal transducer and activator of transcription 3 (STAT3). Results C6 cells treated with 10 and 50 nmol/L TCDD for 24 h showed significant promotion of the proliferation of. The exposure to TCDD resulted in the upregulation in the expression levels of phosphorylated protein kinase B (p-Akt), phosphorylated STAT3, and cyclin D1 in a dose-and time-dependent manner. The inhibition of Akt expression with LY294002 or STAT3 expression with AG490 abolished the TCDD-induced cyclin D1 upregulation and cell proliferation. Furthermore, LY294002 suppressed the activation of STAT3. Finally, TCDD promoted the translocation of STAT3 from the cytoplasm to the nucleus, and LY294002 treatment blocked this effect. Conclusion TCDD exposure promotes the proliferation of astrocyte cells via the Akt/STAT3/cyclin D1 pathway, leading to astrogliosis. -
Key words:
- 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) /
- Akt /
- STAT3 /
- Cyclin D1 /
- Proliferation /
- Astrocytes
-
Figure 1. TCDD induces proliferation of C6 cells. (A) C6 cells were treated with DMSO or 0.1, 10, and 50 nmol/L TCDD for 24 h and subjected to EdU-based proliferation assay. (B) Quantitative analysis of the percentage of EdU-positive cells after TCDD treatment. (C) Flow cytometry analysis was performed to examine the cell cycle phase of C6 glioma cells after treatment with 0, 0.1, 10, and 50 nmol/L TCDD for 24 h. Scale bars: 50 μm. (*P < 0.05, significantly different from the control group).
Figure 2. TCDD activates Akt and STAT3 and increases cyclin D1 expression. (A) The protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH in C6 cells after exposure to different doses of TCDD for 24 h. (B) Quantitative analysis of protein expression relative to GAPDH expression for (A). (C) The protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH in C6 cells after exposure to 10 nmol/L TCDD for indicated time periods. (D) Quantitative analysis of protein expression relative to GAPDH expression for (C). (E) Immunofluorescence staining of STAT3. Primary astrocytes were treated with either 0.1% DMSO (a-c; controls) or 10 nmol/L TCDD (d-f) for 24 h. Nucleus was stained with Hoechst (blue fluorescence); STAT3 was stained with green fluorescence. Scale bars: 25 μm. (*P < 0.05, significantly different from the DMSO-treated group).
Figure 3. Akt/STAT3/cyclin D1 pathway is activated after TCDD treatment. (A) C6 cells were pre-treated with Akt inhibitor LY294002 (50 μmol/L) for 1 h prior to TCDD stimulation. Protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH were analyzed with western blotting. (B) Quantitative analysis of protein expression relative to GAPDH expression for (A). (C) C6 cells were pre-treated with STAT3 inhibitor AG490 (50 μmol/L) for 1 h prior to TCDD stimulation. Protein levels of p-Akt, Akt, p-STAT3, STAT3, cyclin D1, and GAPDH were analyzed with western blotting. (D) Quantitative analysis of protein expression relative to GAPDH expression for (C). (E) Primary astrocytes were treated with 0.1% DMSO (a-c; controls), 10 nmol/L TCDD (d-f), LY294002 (g-i), and LY294002 and TCDD (j-l) for 24 h. Nucleus was stained with Hoechst (blue fluorescence), and STAT3 was stained with green fluorescence. (F) C6 cells were treated with TCDD as indicated in (E), and STAT3 expression in the nucleus or cytoplasm was detected with western blotting. Scale bars: 25 μm. (*P < 0.05, significantly different from the control; #P < 0.05, significantly different from TCDD only group).
Figure 4. The Akt/STAT3 pathway is involved in the TCDD-mediated proliferation of C6 cells. (A) EdU-based proliferation assay was performed to estimate the proliferation rate of C6 cells. Cells were pre-treated with an Akt inhibitor LY294002 (50 μmol/L) or a STAT3 inhibitor AG490 (50 μmol/L) for 1 h prior to TCDD stimulation. (B) Quantitative analysis of EdU-positive cells after TCDD treatment. (C) Flow cytometry analysis showed the cell cycle phase of C6 cells after treatment with 0.1% DMSO, 10 nmol/L TCDD, LY294002 and TCDD, and AG490 and TCDD for 24 h. Scale bars: 50 μm. (*P < 0.05, significantly different from the control group; #P < 0.05, significantly different from TCDD only group).
-
[1] M Zielinski, J Kaminska, M Czerska, et al. Levels and sources of PCDDs, PCDFs and dl-PCBs in the water ecosystems of central Poland-a mini review. Int J Occup Med Environ Health, 2014; 27, 902-18. doi: 10.2478/s13382-014-0336-y [2] D Pelclova, P Urban, Z Fenclova, et al. Neurological and Neurophysiological Findings in Workers with Chronic 2, 3, 7, 8-Tetrachlorodibenzo-p-Dioxin Intoxication 50 Years After Exposure. Basic Clin Pharmacol Toxicol, 2018; 122, 271-7. doi: 10.1111/bcpt.2018.122.issue-2 [3] Wan C, Zhang Y, Jiang J, et al. Critical Role of TAK1-Dependent Nuclear Factor-kappaB Signaling in 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin-induced Astrocyte Activation and Subsequent Neuronal Death. Neurochem Res, 2015; 40, 1220-31. doi: 10.1007/s11064-015-1585-2 [4] Xu G, Zhou Q, Wan C, et al. 2, 3, 7, 8-TCDD induces neurotoxicity and neuronal apoptosis in the rat brain cortex and PC12 cell line through the down-regulation of the Wnt/beta-catenin signaling pathway. Neurotoxicology, 2013; 37, 63-73. doi: 10.1016/j.neuro.2013.04.005 [5] Xu G, Li Y, Yoshimoto K, et al. 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin stimulates proliferation of HAPI microglia by affecting the Akt/GSK-3beta/cyclin D1 signaling pathway. Toxicol Lett, 2014; 224, 362-70. doi: 10.1016/j.toxlet.2013.11.003 [6] C Acosta, H Anderson, C Anderson. Astrocyte dysfunction in Alzheimer disease. J Neurosci Res, 2017; 95, 2430-47. doi: 10.1002/jnr.v95.12 [7] M Pehar, B Harlan, K Killoy, et al. Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Curr Pharm Des, 2017; 23, 5010-21. doi: 10.1016-j.nurt.2010.05.012/ [8] K Panickar, M Norenberg. Astrocytes in cerebral ischemic injury:morphological and general considerations. Glia, 2005; 50, 287-98. doi: 10.1002/(ISSN)1098-1136 [9] L Osborn, W Kamphuis, W Wadman, et al. Astrogliosis:An integral player in the pathogenesis of Alzheimer's disease. Prog Neurobiol, 2016; 144, 121-41. doi: 10.1016/j.pneurobio.2016.01.001 [10] M Sofroniew. Astrogliosis. Cold Spring Harb Perspect Biol, 2014; 7, a020420. http://d.old.wanfangdata.com.cn/Periodical/gwyx-wlyxykf201103012 [11] Zhang Y, Nie X, Tao T, et al. 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin promotes astrocyte activation and the secretion of tumor necrosis factor-alpha via PKC/SSeCKS-dependent mechanisms. J Neurochem, 2014; 129, 839-49. doi: 10.1111/jnc.12696 [12] M Yamaguchi, O Hankinson. 2, 3, 7, 8-Tetrachlorodibenzopdioxin suppresses the growth of human liver cancer HepG2 cells in vitro:Involvement of cell signaling factors. Int J Oncol, 2018; 53, 1657-66. [13] Li Y, Wang K, Jiang Y, et al. 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin (TCDD) inhibits human ovarian cancer cell proliferation. Cell Oncol (Dordr), 2014; 37, 429-37. http://d.old.wanfangdata.com.cn/NSTLQK/NSTL_QKJJ0233938214/ [14] Jin D, J Jung, Y Lee, et al. 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin inhibits cell proliferation through arylhydrocarbon receptor-mediated G1 arrest in SK-N-SH human neuronal cells. Neurosci Lett, 2004; 363, 69-72. doi: 10.1016/j.neulet.2004.03.047 [15] J Herrmann, T Imura, B Song, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci, 2008; 28, 7231-43. doi: 10.1523/JNEUROSCI.1709-08.2008 [16] Zhou H, Yang X, Cui H, et al. Leukemia Inhibitory Factor Contributes to Reactive Astrogliosis via Activation of Signal Transducer and Activator of Transcription 3 Signaling after Intracerebral Hemorrhage in Rats. J Neurotrauma, 2017; 34, 1658-65. doi: 10.1089/neu.2016.4711 [17] Qin A, Yu Q, Gao Y, et al. Inhibition of STAT3/cyclinD1 pathway promotes chemotherapeutic sensitivity of colorectal caner. Biochem Biophys Res Commun, 2015; 457, 681-7. doi: 10.1016/j.bbrc.2015.01.048 [18] R Seiler, G Thalmann, D Rotzer, et al. CCND1/CyclinD1 status in metastasizing bladder cancer:a prognosticator and predictor of chemotherapeutic response. Mod Pathol, 2014; 27, 87-95. doi: 10.1038/modpathol.2013.125 [19] B Manning, L Cantley. AKT/PKB signaling:navigating downstream. Cell, 2007; 129, 1261-74. doi: 10.1016/j.cell.2007.06.009 [20] D Malanga, C De Marco, I Guerriero, et al. The Akt1/IL-6/STAT3 pathway regulates growth of lung tumor initiating cells. Oncotarget, 2015; 6, 42667-86. http://cn.bing.com/academic/profile?id=44254150afbee1f83f1dd3cd2a577c04&encoded=0&v=paper_preview&mkt=zh-cn [21] Chen R, Siao S, Hsu C, et al. TCDD promotes lung tumors via attenuation of apoptosis through activation of the Akt and ERK1/2 signaling pathways. PLoS One, 2014; 9, e99586. doi: 10.1371/journal.pone.0099586 [22] J Davis, F Lauer, A Burdick, et al. Prevention of apoptosis by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) in the MCF-10A cell line:correlation with increased transforming growth factor alpha production. Cancer Res, 2001; 61, 3314-20. http://cn.bing.com/academic/profile?id=7cf42dd3c70960dd77a904c3870ed4e2&encoded=0&v=paper_preview&mkt=zh-cn [23] Zhao J, Zhang Y, Wang C, et al. 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin exposure influence the expression of glutamate transporter GLT-1 in C6 glioma cells via the Ca2+/protein kinase C pathway. J Appl Toxicol, 2016; 36, 1409-17. doi: 10.1002/jat.v36.11 [24] Zhao X, Jin Y, Yang L, et al. Promotion of SIRT1 protein degradation and lower SIRT1 gene expression via reactive oxygen species is involved in Sb-induced apoptosis in BEAS-2b cells. Toxicol Lett, 2018; 296, 73-81. doi: 10.1016/j.toxlet.2018.07.047 [25] L Haim, K Ceyzeriat, M Sauvage, et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer's and Huntington's diseases. J Neurosci, 2015; 35, 2817-29. doi: 10.1523/JNEUROSCI.3516-14.2015 [26] M LeComte, I Shimada, C Sherwin, et al. Notch1-STAT3-ETBR signaling axis controls reactive astrocyte proliferation after brain injury. Proc Natl Acad Sci USA, 2015; 112, 8726-31. doi: 10.1073/pnas.1501029112 [27] M Shiratori-Hayashi, K Koga, H Tozaki-Saitoh, et al. STAT3-dependent reactive astrogliosis in the spinal dorsal horn underlies chronic itch. Nat Med, 2015; 21, 927-31. doi: 10.1038/nm.3912 [28] S Hwang, Y Hwang, M Yun, et al. Indoxyl 3-sulfate stimulates Th17 differentiation enhancing phosphorylation of c-Src and STAT3 to worsen experimental autoimmune encephalomyelitis. Toxicol Lett, 2013; 220, 109-17. doi: 10.1016/j.toxlet.2013.04.016 [29] Wang Y, Chen H, Wang N, et al. Combined 17beta-Estradiol with TCDD Promotes M2 Polarization of Macrophages in the Endometriotic Milieu with Aid of the Interaction between Endometrial Stromal Cells and Macrophages. PLos One, 2015; 10, e0125559. doi: 10.1371/journal.pone.0125559 [30] Li Y, Guo G, Song J, et al. B7-H3 Promotes the Migration and Invasion of Human Bladder Cancer Cells via the PI3K/Akt/STAT3 Signaling Pathway. J Cancer, 2017; 8, 816-24. doi: 10.7150/jca.17759 [31] Guo Y, Zang Y, Lv L, et al. IL8 promotes proliferation and inhibition of apoptosis via STAT3/AKT/NFkappaB pathway in prostate cancer. Mol Med Rep, 2017; 16, 9035-42. doi: 10.3892/mmr.2017.7747 [32] Han R, Zhang F, Wan C, et al. Effect of perfluorooctane sulphonate-induced Kupffer cell activation on hepatocyte proliferation through the NF-kappaB/TNF-alpha/IL-6-dependent pathway. Chemosphere, 2018; 200, 283-94. doi: 10.1016/j.chemosphere.2018.02.137 [33] D Guttridge, C Albanese, J Reuther, et al. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol, 1999; 19, 5785-99. doi: 10.1128/MCB.19.8.5785 [34] Wan C, Liu J, Nie X, et al. 2, 3, 7, 8-Tetrachlorodibenzo-P-dioxin (TCDD) induces premature senescence in human and rodent neuronal cells via ROS-dependent mechanisms. PLos One, 2014; 9, e89811. doi: 10.1371/journal.pone.0089811 [35] Nie X, Liang L, Xi H, et al. 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin induces premature senescence of astrocytes via WNT/beta-catenin signaling and ROS production. J Appl Toxicol, 2015; 35, 851-60. doi: 10.1002/jat.v35.7 [36] Zhao J, Tang C, Nie X, et al. Autophagy potentially protects against 2, 3, 7, 8-tetrachlorodibenzo-p-Dioxin induced apoptosis in SH-SY5Y cells. Environ Toxicol, 2016; 31, 1068-79. doi: 10.1002/tox.v31.9 [37] J Moloney, T Cotter. ROS signalling in the biology of cancer. Semin Cell Dev Biol, 2018; 80, 50-64. doi: 10.1016/j.semcdb.2017.05.023 [38] Wang X, Liu J, Hu J, et al. ROS-activated p38 MAPK/ERK-Akt cascade plays a central role in palmitic acid-stimulated hepatocyte proliferation. Free Radic Biol Med, 2011; 51, 539-51. doi: 10.1016/j.freeradbiomed.2011.04.019 [39] Zhang Z, Duan Q, Zhao H, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-kappaB/STAT3 signaling cascade. Cancer Lett, 2016; 382, 53-63. doi: 10.1016/j.canlet.2016.08.023 [40] C Reale, I Porreca, F Russo, et al. Genetic background and window of exposure contribute to thyroid dysfunction promoted by low-dose exposure to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin in mice. Sci Rep, 2018; 8, 16324. doi: 10.1038/s41598-018-34427-2 [41] O Sorg, M Zennegg, P Schmid, et al. 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko:identification and measurement of TCDD metabolites. Lancet, 2009; 374, 1179-85. doi: 10.1016/S0140-6736(09)60912-0 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2837
- HTML全文浏览量: 705
- PDF下载量: 92
- 被引次数: 0

 Quick Links
Quick Links