

 下载:
下载:


-
Prostate cancer is one of the most common lethal malignancies worldwide[1, 2]. It is initially androgen-dependent but progresses to become a more invasive and androgen- independent, developing resistance to treatment, including chemotherapy and radiotherapy, which ultimately renders it lethal[3, 4]. Therefore, there is a pressing need for new therapeutics to treat androgen-refractory prostate cancer.
In the past few years, significant progress have been made in oncolytic virus area, especially in cancer virotherapy by inhibiting viral replication of cancer cells selectively[5, 6]. It is important to figure out that Hemagglutinating virus of Japan envelope (HVJ-E) has been determined to be capable of evoking several immune responses against tumors[7, 8] and it may also have the ability to eradicate cancer cells through apoptosis, including human multiple myeloma[9], lung cancer cells[10], and melanoma cells[11]. Multiple mechanisms, including actuating MAPK pathways, inducing the death of receptors and mitochondrial pathways, and down regulating the expression of insulin-like growth factor 1 receptor (IGF-1R)[12], have been put forth as being responsible for HVJ-E-induced cell apoptosis. Yet, the mechanisms through which HVJ-E terminates cancer cells have yet to be elucidated, especially for drug-resistant cancer cells.
Autophagy is defined as a general term for the degradation of 'old' lysosomal proteins and damaged organelles[13], which takes on a pro-survival role by supplying cells with the nutrients required to support metabolism when cells are under stress[14]. However, it has been identified that autophagy not only has pro-survival effects but also can promote cell death[15-17]. Differences between these effects may be attributed to the complex and diverse interplays between autophagy and apoptosis. Although numerous studies indicated that autophagy performed a vital part in determining the fate of cancer cells, more research is needed to construct the intricacies of these interconnections. Recently, it has been reported that autophagy is closely connected with apoptosis, which is mediated by reactive oxygen species (ROS)[18, 19]. However, the role of ROS and autophagy in HVJ-E-induced apoptosis in prostate cancer cells remains unclear.
In this study, we characterized for the first time that HVJ-E induced both apoptosis and autophagy in PC3 cells and that induction of apoptosis and autophagy by HVJ-E is associated with ROS generation. Induction of autophagy in PC3 cells and PC3 transplanted tumors was found to promote apoptosis induced by HVJ-E. These discoveries could offer novel evidence supporting the employment of HVJ-E and autophagy inhibitors or inducers in a new treatment plan for androgen-refractory prostate cancers.
-
GFP-microtubule-associated protein 1 light chain 3 (GFP-LC3) plasmid was a gift from Prof. Songshu Meng (Dalian Medical University, China). Hormone-resistant human prostate cancer cell PC3 cell line was purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China) and preserved in our laboratory. PC3 cells were cultured in dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum. Inactivated Sendai virus was prepared as described previously[20]. Briefly, Sendai virus was inoculated into the allantoic cavity of 10-day-old embryonated chicken eggs. Three days later, the allantoic fluid was harvested and the titer of recovered virus was expressed as hemagglutination unit. An Sendai virus suspension of 1.5 × 109 virus particles was inactivated by UV irradiation (198 mJ/cm2). The inactivated Sendai virus that was unable to replicate was referred to as HVJ-E.
-
Antibodies against phospho-specific antibodies to Akt, mTOR, p70S6K, Erk1/2, p38, JNK, along with caspase-3, beclin-1 and total Erk1/2, total p38, total JNK, PARP, β-actin, and inhibitors of U0126, SB203580, and SP600125 were purchased from Cell Signaling Technology (Danvers, MA, USA). Z-VAD-FMK, Rapamycin (Rapa), chloroquine (CQ), NAC, polyclonal rabbit anti-LC3 and HRP-conjugated goat anti-rabbit IgG were obtained from Sigma-Aldrich (St. Louis, USA). FITC Annexin V Apoptosis Detection Kit I was obtained from BD Biosciences (San Diego, US).
-
Cell viability was assessed by CCK-8 assay (Beyotime, Shanghai, China). Briefly, cancer cells were seeded into 96-well plates at 1 × 104 cell per well and incubated at 37 ℃ for 24 h. After treatments under various conditions, 10 μL of kit reagent was added to cells followed by incubation for 4 h at 37 ℃. Then optical density (OD) value were read at 450 nm using a Bio-Teck ELISA microplate reader. Viability was calculated using the ratio of OD in the experimental wells to those in wells with cells grown in medium or control cells. For the clonogenic assay, 1, 000 cells were seeded into 60 mm dishes in triplicate, treated with HVJ-E at 0, 100, 400, and 800 multiplicity of infection (MOI), followed by incubation for 9 days. Colonies were fixed, stained, and counted under an inverted microscope. Colonies with 50 cells or more were counted.
-
Intracellular ROS were measured using 2', 7'-dichlorofluorescein diacetate (DCFH-DA, Beyotime, Shanghai, China). A total of 1 × 105 PC3 cells were either mock-treated (PBS) or treated with HVJ-E at a MOI of 800 for 24 h. To verify if ROS generation is related to HVJ-E-induced cellular apoptosis, some cells were treated with NAC, an ROS scavenger. To perform this assay, PC3 cells were incubated with NAC (100 μmol/L) for 30 min prior to HVJ-E treatment. Collected cells were analyzed by flow cytometry (Becton-Dickinson, San Jose, CA) in quadruplicate.
-
Apoptosis was measured by flow cytometry using an Annexin V and propidium iodide (PI) double-staining technique, as described[21]. An aggregate of 4 × 105 PC3 cells were mock-treated or treated with HVJ-E at a MOI of 100, 400, or 800 for 24 h. Relative levels of apoptosis were measured. With the apoptosis inhibition assay, cells were incubated with a Z-VAD-FMK inhibitor (40 μmol/L), CQ (7.5 μmol/L), or Rapa (100 nmol/L) for 30 min before being treated with HVJ-E (800 MOI). Apoptosis was measured in HVJ-E-treated cells at the indicated MOI.
-
Lipofectamine 2, 000 was used to transfect PC3 cells with plasmid GFP-LC3 according to the manufacturer's instructions. Dot formation of GFP-LC3 was observed under a fluorescent microscope (DMI 3000B, Leica) after drug or HVJ-E treatment. An aggregate of 100 transfected cells were evaluated in every well for the 3 separate experiments.
-
Cells placed in 60 mm-dishes were treated with HVJ-E at different MOI. After 24 h treatment, cells were gathered and lysed. An identical quantity of protein (30 μg) was divided via 10%-15% SDS-PAGE and moved to NC membranes. After blocking with 5% non-fat milk for 2 h, the membrane was incubated with primary antibodies against the target proteins at 4 ℃ overnight. Immunoreactive bands were viewed via enhanced chemiluminescence (ECL) using HRP-conjugated IgG secondary antibodies.
-
Nude male mice were subcutaneously inoculated in the side with PC3 cells (5 × 106 cells in 100 μL PBS per mouse) to prompt tumor growth. Tumor-bearing animals were intratumorally injected with HVJ-E when the diameter of the tumors was approximately 0.5 cm. Mice were indiscriminately devided into 6 groups (7 mice per group): (a) vehicle (PBS) treatment, (b, c) intraperitoneal treatment with Rapa (5 mg/kg) or CQ (40 mg/kg) alone 3 times a week, (d) 3 rounds of intratumoral administration with HVJ-E (5 × 109 virus particles per dose), and (e, f) HVJ-E along with CQ or Rapa (the identical dose as described prior) given 1 d before HVJ-E injection. At 7 d after treatment, 2 mice (of the 7 in each group) were killed, and tumor protein was separated for caspase-3 activation and LC3Ⅰ/Ⅱ abundance evaluation. Meanwhile, the tumor volume of the remaining mice was measured at 3-day intervals with calipers. Tumor volume was calculated based on the following formula: volume = (length) × (width)2 /2[22].
All animal experiments were approved by the Animal Care and Use Committee of Yangzhou University (Approval ID: SYXK (Su) 2012-0029).
-
Significant variance between groups was performed for all groups using a one-way ANOVA. Data are expressed as means ± SE. Multiple comparisons among groups were evaluated using Dunnett's LSD test. Differences with P < 0.05 were considered statistically significant.
-
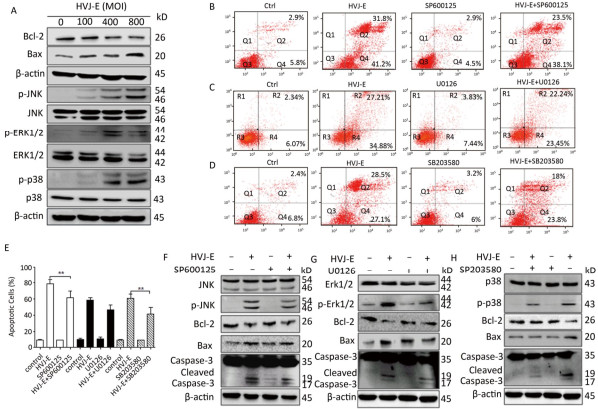
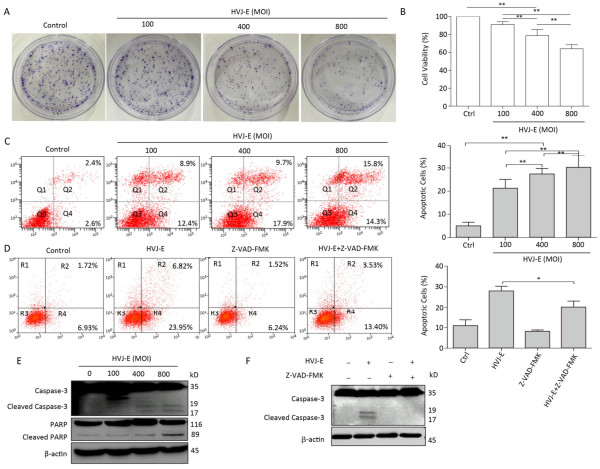
To determine whether HVJ-E had an effect on prostate cancer cell viability, PC3 cells were treated with HVJ-E at the indicated MOI for 24 h, and colony formation and cell viability assays confirmed that HVJ-E dose-dependently reduced cell viability and clonogenic cell survival vs. controls (Figure 1A and 1B), implying that HVJ-E might have an oncolytic effect on cancer cells in the human prostate. After this, Annexin-V/PI double staining at various MOI (100, 400, and 800) was used to ascertain the accountability of reduced proliferation and viability in apoptosis, and apoptosis in PC3 cells was found to respond in a dose-dependent manner to HVJ-E treatment (Figure 1C), further indicating that HVJ-E induced an oncolytic effect on PC3 cells. HVJ-E induced apoptosis was also verified by the observation of cleaved caspase-3 and PARP (Figure 1E).
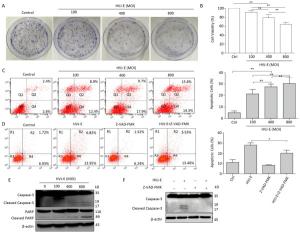
Figure 1. HVJ-E induced apoptosis in PC3 cells relies on caspase. (A) HVJ-E at different MOI limited colony formation in the PC3 cells. (B) The impact of HVJ-E at the noted MOI on PC3 cell viability examined with a CCK-8-based assay. Data were means ± SD of 3 experiments performed in triplicate. **P < 0.01. (C) PC3 cells were mock- or HVJ-E-treated at the MOI of 100, 400, and 800 for 24 h. The cells were gathered and double-stained with Annexin V and PI and then assessed with flow cytometry. Data were means ± SD from 3 independent experiments. **P < 0.01. (D) PC3 cells were mock-treated or pretreated with the pan-caspase inhibitor Z-VAD-FMK (40 μmol/L) prior to HVJ-E treatment (MOI = 800), and apoptotic cells were assessed with flow cytometry. *P < 0.05. (E) Quantification of activated types of caspase-3 and cleaved PARP.β-actin acted as a loading control. (F) Cells were treated as detailed in the METHODS section, and then Western blot was used to evaluate the cleavage of caspase-3.
Cells were pretreated with the pan-caspase inhibitor Z-VAD-FMK (40 μmol/L), and apoptotic cells were assayed via flow cytometry to confirm if caspase is needed for HVJ-E-induced apoptosis within PC3 cells. As shown in Figure 1D, treatment with Z-VAD-FMK for 24 h substantially decreased HVJ-E-induced apoptosis in the PC3 cells. In addition, as revealed in Figure 1F, pretreatment with the inhibitor significantly decreased HVJ-E-induced cleavage of caspase-3 and PARP, which indicated that caspase activity was needed for HVJ-E-induced apoptosis in PC3 cells.
-
MAPK cascades perform vital parts in inducing apoptosis in response to various stresses, including inactivated Sendai virus treatment[23]. To explore the role of MAPK pathway in HVJ-E-induced apoptosis in PC3 cells, a post-HVJ-E-treatment dose-course of Erk1/2, JNK, and p38 MAPK phosphorylation was evaluated. As illustrated in Figure 2A, the phosphorylation of Erk1/2, p38 MAPK, and JNK were increased by HVJ-E while the ratio of Bcl-2/Bax was decreased significantly. To determine whether MAPK pathway was required for HVJ-E-induced apoptosis in PC3 cells, the inhibitors SB203580, SP600125, and U0126 targeting p38 MAPK, JNK, and Erk1/2 respectively were added to cell culture 0.5 h prior to HVJ-E treatment. As shown in Figure 2, SB203580 and SP600125 significantly inhibited HVJ-E-induced apoptosis with 48 h post-treatment (P < 0.01), but U0126 did not significantly inhibit HVJ-E induced apoptosis (Figure 2B-E), which were confirmed by the result of western blot (Figure 2G-H). Above all, these results indicated that JNK and p38 MAPK were involved in HVJ-E-induced apoptosis of PC3 cells.
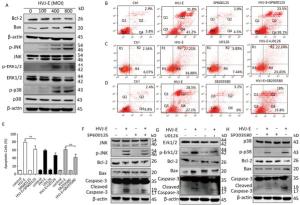
Figure 2. JNK and p38 MAPK pathways took part in HVJ-E-induced apoptosis in PC3 cells. (A) PC3 cells were treated as detailed, and MAPK activation was quantified with particular antibodies against phosphorylated Erk1/2, JNK, and p38. Total protein was quantified as a loading control. (B-E) PC3 cells were mock-treated or pretreated with the subsequent MAPK inhibitors for 30 min: JNK inhibitor SP600125 (2 μmol/L), p38 inhibitor SB203580 (10 μmol/L), and Erk1/2 inhibitor U0126 (4 μmol/L). Next, cells were treated with HVJ-E at 800 MOI for 48 h, and cells were assayed with Flow cytometry. Data are means ± SD of 3 independent experiments, **P < 0.01. (F-H) Cell lysates were set up for Western blot to determine target pathway inactivation.
-
It has been reported that ROS can activate MAPK signaling pathways including JNK, Erk1/2, and p38 MAPK[24]. PC3 cells were treated with HVJ-E at 800 MOI for 24 h to evaluate if HVJ-E prompted ROS production in prostate cancer cells. As demonstrated in Figure 3A, HVJ-E induced significant ROS buildup in PC3 cells. The cells were pretreated with a ROS scavenger, NAC, before HVJ-E to examine the operation of ROS in HVJ-E-induced apoptosis in PC3 cells. The outcomes suggested that in addition to blocking ROS generation (Figure 3A), it also significantly limited HVJ-E-induced apoptosis (Figure 3B). Moreover, the immunoblot assay outcomes indicated that NAC treatment prevented PC3 cells from being subjected to HVJ-E-induced initiation of JNK, p38 MAPK, and caspase-3 (Figure 3C); these outcomes implied that the JNK and p38 MAPK pathways took part in the management of PC3 apoptosis downstream of ROS.
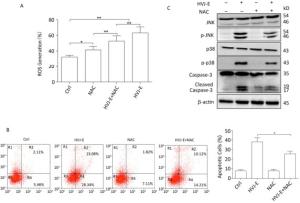
Figure 3. HVJ-E-induced apoptosis in PC3 cells by JNK and p38 MAPK activation was ROS-dependent. (A) Cells were mock-treated or pretreated with NAC (100 μmol/L). Cells were then treated with HVJ-E (MOI = 800). Intracellular ROS was quantified with the DCF-DA method. Data are means ± SD of experiments conducted in quadruplicate. (B) Cells were treated as detailed, and apoptosis was determined by flow cytometry. Data are means ± SDs for 3 experiments. (C) JNK and p38 MAPK activation were evaluated with particular antibodies against phosphorylated JNK and p38. The total protein expression was quantified and implemented as a loading control. *P < 0.05, **P < 0.01.
-
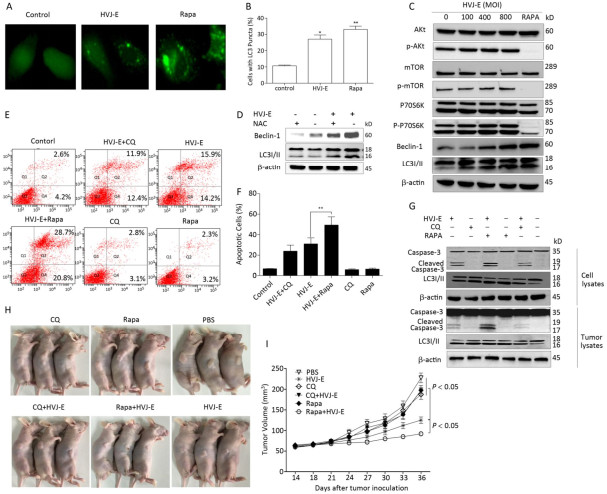
Both autophagy and apoptosis were triggered after treatment with numerous viruses[25]. To determine whether autophagy was induced by HVJ-E, PC3 cells were transfected with plasmid GFP-LC3, followed by HVJ-E (800 MOI) treatment for 24 h. Rapa was a positive control. As shown in Figure 4A and 4B, more bright dots appeared in HVJ-E-treated and Rapa-treated cells than in control cells (P < 0.05). In addition, as shown in Figure 4C, there was far more conversion of endogenous LC3 to LC3Ⅱ in PC3 cells than in control cells. In this way, HVJ-E induced autophagy in PC3 cells.
The class Ⅰ PI3K/Akt/mTOR/p70S6K pathway negatively regulated autophagy, whereas the class Ⅲ PI3K/beclin-1 pathway promoted autophagic induction[26]. Since the two signaling pathways were activatede in the control of apoptosis and autophagy, we established if HVJ-E impacted these pathways. As revealed in Figure 4C, the phosphorylation of Akt, mTOR, and p70S6K with HVJ-E treatment was not impacted; however, beclin-1 expression was initiated upon HVJ-E treatment. These data indicated that the class Ⅲ PI3K/beclin-1 signaling pathway was actuated with HVJ-E treatment and may lead to HVJ-E-triggered autophagy. To determine whether HVJ-E-induced autophagy in PC3 cells is ROS-dependent, cells were treated with NAC prior to HVJ-E treatment; NAC significantly inhibited HVJ-E-induced LC3 conversion and beclin-1 activation (Figure 4D), suggesting that HVJ-E-induced autophagy might depend on ROS generation.
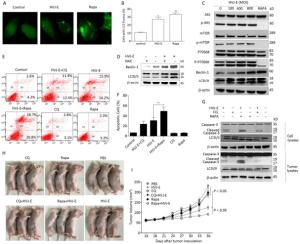
Figure 4. Pharmacological modulation of autophagy improved HVJ-E-induced apoptosis. (A, B) Percentage of cells with typical LC3 bright dots (> 5 bright dots per cell) were shown. Data were means ± SD from 3 separate experiments (a minimum of 100 cells were counted for every experiment, *P < 0.05). (C) HVJ-E induced autophagy in PC3 cells by the initiation of beclin-1. Total and phosphorylated Akt, mTOR, and p70SK and beclin-1 and LC3 expression were measured with Western blot. Total protein was determined as a loading control. (D) Cells were mock-treated or pretreated with NAC (100 μmol/L). The cells were then treated with HVJ-E (MOI = 800), and the expression of beclin-1 and LC3 was determined by Western blot. Total protein was evaluated as the loading control. (E, F) Cells were treated with CQ (7.5 μmol/L) or Rapa (125 nmol/L) for 30 min, and then with HVJ-E (800 MOI) for 24 h. Apoptosis was determined with flow cytometry 24 h following treatment. Data were means ± SD of 3 independent experiments, **P < 0.01. (G) Cells or tumors were treated as detailed, and LC3-Ⅱ conversion and cleavage of caspase-3 was evaluated with Western blot. (H, I) The mice were treated as detailed above. Tumor volumes were evaluated at 3-day intervals over 36 days after injections and expressed as the mean ± SD (n = 5) in tumor volume-time curves. The variation in tumor regression among the HVJ-E-treated and vehicle groups was significant (P < 0.05); the groups that were administered the combined treatment and the single-treatment (just the virus or just the drugs) group (P < 0.05). No statistically significant difference was noted among the groups administered the single treatments and the vehicle-treated group.
-
Both the autophagy inducer Rapa and the inhibitor CQ were implemented in the examination of the interaction between HVJ-E-mediated autophagy and apoptosis in PC3 cells[27]. It has been reported that Rapa induced autophagy by inhibiting mTOR/p70S6K which is MAPK independent. As shown in Figure 4G, pre-treatment of Rapa or CQ to PC3 cells enhanced LC3 Ⅱ accumulation with HVJ-E treatment, enhancing induction of autophagy by Rapa and inhibiting autophagosome-lysosome fusion by CQ in HVJ-E- treated cells.
We then evaluated if pharmacological modulation of autophagy impacted HVJ-E-mediated apoptosis. HVJ-E-mediated apoptosis in Rapa-treated PC3 cells was significantly increased as noted by flow cytometry (Figure 4E and 4F, P < 0.01), substantiating the observation that Rapa (but not CQ) improved the cleavage of caspase-3 (Figure 4G).
We examined the oncolytic impacts of HVJ-E with Rapa or CQ in PC3 cell-bearing mice to confirm the therapeutic importance of autophagy modulators and HVJ-E. Tumor-bearing mice were intraperitoneally injected with the vehicle, Rapa, or CQ and administered HVJ-E intratumorally after 24 h. Evaluations of caspase-3 activation in PC3-derived tumors showed that pretreatment with Rapa produced stronger caspase-3 activation in contrast to the tumors with just HVJ-E treatment (Figure 4G). The cleaved caspase-3 levels were hardly noticeable in vehicle-, CQ-, and Rapa-treated tumors. We then determined if the in vivo combination treatments led to increased inhibition of tumor cell development as shown in the in vitro experiments. The treatment of PC3 cell-bearing tumors with just Rapa or Rapa and CQ had insignificant therapeutic impact on tumor development (Figure 4H and 4I). As expected, HVJ-E therapy markedly reduced tumor growth relative to vehicle treatment (Figure 4H and 4I, P < 0.05, respectively). More importantly, combination treatment with HVJ-E with Rapa resulted in significantly more inhibition of tumor growth than HVJ-E alone (Figure 4H and 4I). Taken together, these outcomes imply that autophagy may operate as a death mechanism in HVJ-E-treated PC3 cells.
-
Many studies suggested that HVJ-E may have a therapeutic effect in cancer treatment, but the manner by which this occurs is unclear. A current study detailed HVJ-E-induced cell termination in human neuroblastoma cells as relying on necrosis instead of apoptosis[28]. Yet, the research we performed previously indicated that HVJ-E-induced cell death depends on apoptosis via caspase-8 or -9 or activation of both in cancer cells[13, 23]. Differences in cell lines may contribute to the diversity of caspase activation after HVJ-E treatment as well. The present experiments revealed that HVJ-E had significant apoptotic characteristics in PC3 cells, and these data are in agreement with outcomes acquired from different oncolytic viruses[27]. Further, HVJ-E-induced apoptosis was inhibited by the pan-caspase inhibitor, Z-VAD-FMK, validating that the initiation of cell death relied on caspase-mediated apoptotic pathways.
MAPK signal pathways have been acknowledged to take on crucial roles in the management of cell proliferation and apoptosis in reaction to different stimuli[21]. To the best of our knowledge, a limited number of reports have been published concerning activation of MAPK pathways in HVJ-E-induced apoptosis[10, 23], but the manner in which MAPK pathways are activated after HVJ-E treatment are unclear. Thus, we suggest that HVJ-E treatment activated all 3 major MAPK pathways in PC3 cells, and this is consistent with our previous study[23]. However, results of MAPK pathway inhibition experiments verified that JNK and p38 MAPK pathways were associated with HVJ-E-induced cytotoxicity in PC3 cells. This is the first work to demonstrate that HVJ-E-induced ROS may be an inducer of JNK and p38 MAPK activation. When ROS scavenger NAC was employed before HVJ-E treatment, it obstructed ROS generation and it also significantly limited HVJ-E-induced activation of JNK and p38 MAPK and cleavage of caspase-3 and apoptosis.
Additionally to the induction of apoptosis, we here propose another role of HVJ-E, the induction of autophagy in PC3 cells via the activation of class Ⅲ PI3K/beclin-1 pathway, which positively regulates autophagy[29]. We discovered in our prior paper that HVJ-E induced the autophagic operation in A549 and A549/DDP cells by limiting the class Ⅰ PI3K/Akt/mTOR/p70S6K negative regulatory pathway[10]. Interestingly, we also found that HVJ-E-induced autophagy in PC3 cells depends partially on HVJ-E-induced ROS, and these results are consistent with those data, suggesting that silibinin could induce ROS and apoptosis in human prostate cancer PC-3 cells[25].
We then examined the role of autophagy in HVJ-E-induced apoptosis. Pharmacological autophagy modulators, including Rapa and CQ, have been employed along with oncolytic virus to improve the therapeutic impact of treating different types of cancers in preclinical trials[30, 31]. Here, we present both in vitro and in vivo evidence that Rapa enhances HVJ-E-mediated apoptosis in PC3 cells, but CQ did not change anticancer effects of HVJ-E in PC3 cells. Augmentation in HVJ-E-mediated apoptosis in the presence of autophagy modulators were also supported by activation of caspase-3. Interestingly, Kim's group reported that the autophagy inhibitor 3-Methyladenine (3-MA) augmented the anticancer effects of silibinin in PC3 cells, which is not consistent with our results[25]. The implementation of autophagy inducers or inhibitors in combination cancer treatments could be virus strain- and drug-dependent.
Overall, HVJ-E regulated PI3K/beclin-1 and JNK and p38 pathway in prostate cancer cells, and this was ROS-dependent, thereby modulating autophagy and apoptosis. Induction of autophagy induced by HVJ-E in PC3 cells promoted apoptosis. These outcomes offer new knowledge about the latent mechanism of the anti-tumor effects of HVJ-E and furnish further understanding of different ways to diminish or eliminate human cancers.
doi: 10.3967/bes2018.036
Inactivated Sendai Virus Induces ROS-dependent Apoptosis and Autophagy in Human Prostate Cancer Cells
-
Abstract:
Objective The current study aims to investigate the effect of Hemagglutinating virus of Japan envelope (HVJ-E) on induction of apoptosis and autophagy in human prostate cancer PC3 cells, and the underlying mechanisms. Methods PC3 cells were treated with HVJ-E at various multiplicity of infection (MOI), and the generated reactive oxygen species (ROS), cell viability, apoptosis, and autophagy were detected, respectively. Next, the role of ROS played in the regulation of HVJ-E-induced apoptosis and autuphagy in PC3 cells were analysed. In the end, the relationship between HVJ-E-induced apoptosis and autuophagy was investigated by using rapamycin and chloroquine. Results Flow cytometry assay revealed that HVJ-E treatment induced dose-dependent apoptosis and that the JNK and p38 MAPK signaling pathways were involved in HVJ-E-induced apoptosis in PC3 cells. In addition, HVJ-E was able to induce autophagy in PC3 cells via the class Ⅲ PI3K/beclin-1 pathway. The data also implyed that HVJ-E-triggered autophagy and apoptosis were ROS dependent. When ROS was blocked with N-acetylcysteine (NAC), HVJ-E-induced LC3-Ⅱ conversion and apoptosis were reversed. Interestingly, HVJ-E-induced apoptosis was significantly increased by an inducer of autophagy, rapamycin pretreatment, both in vitro and in vivo. Conclusion HVJ-E exerts anticancer effects via autophagic cell death in prostate cancer cells. -
Key words:
- Inactivated Sendai virus (HVJ-E) /
- Reactive oxygen species (ROS) /
- Apoptosis /
- Autophagy
-
Figure 1. HVJ-E induced apoptosis in PC3 cells relies on caspase. (A) HVJ-E at different MOI limited colony formation in the PC3 cells. (B) The impact of HVJ-E at the noted MOI on PC3 cell viability examined with a CCK-8-based assay. Data were means ± SD of 3 experiments performed in triplicate. **P < 0.01. (C) PC3 cells were mock- or HVJ-E-treated at the MOI of 100, 400, and 800 for 24 h. The cells were gathered and double-stained with Annexin V and PI and then assessed with flow cytometry. Data were means ± SD from 3 independent experiments. **P < 0.01. (D) PC3 cells were mock-treated or pretreated with the pan-caspase inhibitor Z-VAD-FMK (40 μmol/L) prior to HVJ-E treatment (MOI = 800), and apoptotic cells were assessed with flow cytometry. *P < 0.05. (E) Quantification of activated types of caspase-3 and cleaved PARP.β-actin acted as a loading control. (F) Cells were treated as detailed in the METHODS section, and then Western blot was used to evaluate the cleavage of caspase-3.
Figure 2. JNK and p38 MAPK pathways took part in HVJ-E-induced apoptosis in PC3 cells. (A) PC3 cells were treated as detailed, and MAPK activation was quantified with particular antibodies against phosphorylated Erk1/2, JNK, and p38. Total protein was quantified as a loading control. (B-E) PC3 cells were mock-treated or pretreated with the subsequent MAPK inhibitors for 30 min: JNK inhibitor SP600125 (2 μmol/L), p38 inhibitor SB203580 (10 μmol/L), and Erk1/2 inhibitor U0126 (4 μmol/L). Next, cells were treated with HVJ-E at 800 MOI for 48 h, and cells were assayed with Flow cytometry. Data are means ± SD of 3 independent experiments, **P < 0.01. (F-H) Cell lysates were set up for Western blot to determine target pathway inactivation.
Figure 3. HVJ-E-induced apoptosis in PC3 cells by JNK and p38 MAPK activation was ROS-dependent. (A) Cells were mock-treated or pretreated with NAC (100 μmol/L). Cells were then treated with HVJ-E (MOI = 800). Intracellular ROS was quantified with the DCF-DA method. Data are means ± SD of experiments conducted in quadruplicate. (B) Cells were treated as detailed, and apoptosis was determined by flow cytometry. Data are means ± SDs for 3 experiments. (C) JNK and p38 MAPK activation were evaluated with particular antibodies against phosphorylated JNK and p38. The total protein expression was quantified and implemented as a loading control. *P < 0.05, **P < 0.01.
Figure 4. Pharmacological modulation of autophagy improved HVJ-E-induced apoptosis. (A, B) Percentage of cells with typical LC3 bright dots (> 5 bright dots per cell) were shown. Data were means ± SD from 3 separate experiments (a minimum of 100 cells were counted for every experiment, *P < 0.05). (C) HVJ-E induced autophagy in PC3 cells by the initiation of beclin-1. Total and phosphorylated Akt, mTOR, and p70SK and beclin-1 and LC3 expression were measured with Western blot. Total protein was determined as a loading control. (D) Cells were mock-treated or pretreated with NAC (100 μmol/L). The cells were then treated with HVJ-E (MOI = 800), and the expression of beclin-1 and LC3 was determined by Western blot. Total protein was evaluated as the loading control. (E, F) Cells were treated with CQ (7.5 μmol/L) or Rapa (125 nmol/L) for 30 min, and then with HVJ-E (800 MOI) for 24 h. Apoptosis was determined with flow cytometry 24 h following treatment. Data were means ± SD of 3 independent experiments, **P < 0.01. (G) Cells or tumors were treated as detailed, and LC3-Ⅱ conversion and cleavage of caspase-3 was evaluated with Western blot. (H, I) The mice were treated as detailed above. Tumor volumes were evaluated at 3-day intervals over 36 days after injections and expressed as the mean ± SD (n = 5) in tumor volume-time curves. The variation in tumor regression among the HVJ-E-treated and vehicle groups was significant (P < 0.05); the groups that were administered the combined treatment and the single-treatment (just the virus or just the drugs) group (P < 0.05). No statistically significant difference was noted among the groups administered the single treatments and the vehicle-treated group.
-
[1] Chen M, Zhang J, Sampieri K, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet, 2018; 50, 206-18. doi: 10.1038/s41588-017-0027-2 [2] Goodall J, Mateo J, Yuan W, et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov, 2017; 7, 1006-17. doi: 10.1158/2159-8290.CD-17-0261 [3] Liang C, Wang S, Qin C, et al. TRIM36, a novel androgen-responsive gene, enhances anti-androgen efficacy against prostate cancer by inhibiting MAPK/ERK signaling pathways. Cell Death Dis, 2018; 9, 155. doi: 10.1038/s41419-017-0197-y [4] Ghasemi S, Lorigooini Z, Wibowo JP, et al. Tricin isolated from Allium atroviolaceum potentiated the effect of docetaxel on PC3 cell proliferation: role of miR-21. Nat Prod Res, 2018; 1-4. https://www.researchgate.net/publication/282729087_Isolation_and_structure_identification_of_flavonoids_from_leaves_of_Zea_mays_L [5] Meng S, Zhou Z, Chen F, et al. Newcastle disease virus induces apoptosis in cisplatin-resistant human lung adenocarcinoma A549 cells in vitro and in vivo. Cancer lett, 2012; 317, 56-64. doi: 10.1016/j.canlet.2011.11.008 [6] Yoo SY, Bang SY, Jeong SN, et al. A cancer-favoring oncolytic vaccinia virus shows enhanced suppression of stem-cell like colon cancer. Oncotarget, 2016; 7, 16479-89. http://cn.bing.com/academic/profile?id=839a9ef1b9abb17ed67f94d32c6f441b&encoded=0&v=paper_preview&mkt=zh-cn [7] Li S, Nishikawa T, Kaneda Y. Inactivated Sendai virus particle upregulates cancer cell expression of intercellular adhesion molecule-1 and enhances natural killer cell sensitivity on cancer cells. Cancer Sci, 2017; 108, 2333-41. doi: 10.1111/cas.2017.108.issue-12 [8] Fujihara A, Kurooka M, Miki T, et al. Intratumoral injection of inactivated Sendai virus particles elicits strong antitumor activity by enhancing local CXCL10 expression and systemic NK cell activation. Cancer Immunol Immun, 2008; 57, 73-84. doi: 10.1007/s00262-007-0351-y [9] Jiang Y, Saga K, Miyamoto Y, et al. Cytoplasmic calcium increase via fusion with inactivated Sendai virus induces apoptosis in human multiple myeloma cells by downregulation of c-Myc oncogene. Oncotarget, 2016; 24, 36034-48. http://cn.bing.com/academic/profile?id=07ee6ca197c79f6bfbc6efd5419dfa1f&encoded=0&v=paper_preview&mkt=zh-cn [10] Zhang Q, Zhu H, Xu X, et al. Inactivated Sendai virus induces apoptosis and autophagy via the PI3K/Akt/mTOR/p70S6K pathway in human non-small cell lung cancer cells. Biochem Bioph Res Co, 2015; 465, 64-70. doi: 10.1016/j.bbrc.2015.07.130 [11] Nishikawa T, Tung LY, Kaneda Y. Systemic administration of platelets incorporating inactivated Sendai virus eradicates melanoma in mice. Mol Ther, 2014; 22, 2046-55. doi: 10.1038/mt.2014.128 [12] Gao H, Xu XS, Chen ZD, et al. Inactivated sendai virus induces apoptosis in murine melanoma cells by IGF-1R down-regulation. Biomed Environ Sci, 2013; 26, 998-1002. http://cn.bing.com/academic/profile?id=9b1f0b1e584c20862c6a4d3e731a7a3f&encoded=0&v=paper_preview&mkt=zh-cn [13] Yue H, Li W, Liu P, et al. Inhibition of autophagy promoted sphingosylphosphorylcholine induced cell death in nonsmall cell lung cancer cells. Biochem Bioph Res Co, 2014; 453, 502-7. doi: 10.1016/j.bbrc.2014.09.120 [14] Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell metab, 2017; 25, 1037-43. doi: 10.1016/j.cmet.2017.04.004 [15] Bo Q, Ma S, Han Q, et al. Role of autophagy in photoreceptor cell survival and death. Crit Rev Eukar Gene, 2015; 25, 23-32. doi: 10.1615/CritRevEukaryotGeneExpr.v25.i1 [16] Castino R, Isidoro C, Murphy D. Autophagy-dependent cell survival and cell death in an autosomal dominant familial neurohypophyseal diabetes insipidus in vitro model. FASEB J, 2005; 19, 1024-6. doi: 10.1096/fj.04-3163fje [17] Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ, 2005; 12, 1509-18. doi: 10.1038/sj.cdd.4401751 [18] Koul M, Kumar A, Deshidi R, et al. Cladosporol A triggers apoptosis sensitivity by ROS-mediated autophagic flux in human breast cancer cells. BMC cell Biol, 2017; 18, 26. doi: 10.1186/s12860-017-0141-0 [19] Li R, Zhou P, Guo Y, et al. Tris (1, 3-dichloro-2-propyl) phosphate induces apoptosis and autophagy in SH-SY5Y cells: Involvement of ROS-mediated AMPK/mTOR/ULK1 pathways. Food Chem Toxicol, 2017; 100, 183-96. doi: 10.1016/j.fct.2016.12.029 [20] Zhang Q, Wang Z, Yuan Y, et al. Immunoadjuvant effects of hemagglutinating virus of Japan envelope (HVJ-E) on the inactivated H9 subtype avian influenza virus vaccine. Vet Immunol Immunopathol, 2011; 141, 116-23. doi: 10.1016/j.vetimm.2011.02.021 [21] Bian J, Wang K, Kong X, et al. Caspase- and p38-MAPK-dependent induction of apoptosis in A549 lung cancer cells by Newcastle disease virus. Arch Virol, 2011; 156, 1335-44. doi: 10.1007/s00705-011-0987-y [22] Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemoth Pharm, 1989; 24, 148-54. doi: 10.1007/BF00300234 [23] Zhang Q, Xu X, Yuan Y, et al. IPS-1 plays a dual function to directly induce apoptosis in murine melanoma cells by inactivated Sendai virus. Int J Cancer, 2014; 134, 224-34. doi: 10.1002/ijc.v134.1 [24] McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Sign, 2006; 8, 1775-89. doi: 10.1089/ars.2006.8.1775 [25] Kim SH, Kim KY, Yu SN, et al. Autophagy inhibition enhances silibinin-induced apoptosis by regulating reactive oxygen species production in human prostate cancer PC-3 cells. Biochem Bioph Res Co, 2015; 468, 151-6. doi: 10.1016/j.bbrc.2015.10.143 [26] Myers R, Coviello C, Erbs P, et al. Polymeric Cups for Cavitationmediated Delivery of Oncolytic Vaccinia Virus. Mol Ther, 2016; 24, 1627-33. doi: 10.1038/mt.2016.139 [27] Mansfield DC, Kyula JN, Rosenfelder N, et al. Oncolytic vaccinia virus as a vector for therapeutic sodium iodide symporter gene therapy in prostate cancer. Gene ther, 2016; 23, 357-68. doi: 10.1038/gt.2016.5 [28] Nomura M, Ueno A, Saga K, et al. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res, 2014; 74, 1056-66. doi: 10.1158/0008-5472.CAN-13-1283 [29] Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J pathol, 2010; 221, 3-12. doi: 10.1002/path.2697 [30] Fend L, Remy-Ziller C, Foloppe J, et al. Oncolytic virotherapy with an armed vaccinia virus in an orthotopic model of renal carcinoma is associated with modification of the tumor microenvironment. Oncoimmunology, 2016; 5, e1080414. doi: 10.1080/2162402X.2015.1080414 [31] Gholami S, Marano A, Chen NG, et al. Erratum to: A novel vaccinia virus with dual oncolytic and anti-angiogenic therapeutic effects against triple-negative breast cancer. Breast Cancer Res Tr, 2016; 156, 607-8. doi: 10.1007/s10549-016-3767-2 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3350
- HTML全文浏览量: 1042
- PDF下载量: 185
- 被引次数: 0

 Quick Links
Quick Links