

 下载:
下载:

-
Ebola virus disease is an acute hemorrhagic infection caused by Ebola virus[1]. Five Ebola virus subtypes have been identified[2]: Zaire ebolavirus (EBOV), Sudan ebolavirus (SUDV), Reston ebolavirus (RESTV), TaiForest ebolavirus (TAFV), and Bundibugyo ebolavirus (BDBV). EBOV, SUDV, and BDBV can be transmitted from person-to-person. Since the Ebola virus was discovered in 1976[1], there have been many outbreaks. According to the World Health Organization, as of the end of 2012, EBOV had caused 1, 388 confirmed infections with a mortality rate of 79%, SUDV had caused 792 infections with a mortality rate of 54%, and BDBV had caused 206 infections with a mortality rate of 32%[3]. In December 2013, an outbreak occurred in Guinea followed by the rapid spread to several West African countries, including Sierra Leone and Liberia[4]. The epidemic was caused by a new strain of EBOV. There were 28, 652 cases of suspected and confirmed infections, and 11, 325 deaths[5]. Reports of sporadic infections continue.
The development of an economical, easy-to-use preventive vaccine is the most effective means to control the spread of such an epidemic. Since 1980, a number of international research teams have begun to develop EBOV vaccines, including inactivated virus vaccines[6], replication-deficient virus vaccines, adenoviral vector vaccines cAd3-EBOV[7], herpetic stomatitis virus vector vaccines rVSV-ZEBOV, and others[8-11].
EBOV has an envelope structure. A membrane glycoprotein (GP) can mediate virus adsorption[12-13], internalization, and membrane fusion. It has been found that the GP can interact with cell surface receptors without the aid of other viral proteins[14]. GP proteins can induce the host to produce high titers of neutralizing antibodies that protect the host from viral infection[15]. In our previous studies, a recombinant Zaire-type Ebola membrane protein GP-Fc subunit vaccine and a DNA vector vaccine containing the recombinant membrane protein GP-Fc were prepared. The results of mouse immunization showed that both vaccines could stimulate higher levels of specific antibodies[16-17]. However, whether neutralizing antibodies were induced by the two vaccines was not evaluated.
Neutralization trials using EBOV require a biosafety level 4 laboratory (BSL-4), which limits validation testing. The use of pseudoviruses to study antiviral drugs or vaccines is a common method of virological research[18-19]. Therefore, this study used a pseudovirus for neutralization antibody titer detection. Based on the human immunodeficiency virus-1 (HIV-1) virus gene sequence, the vector pNL 4-3.Luc.RE containing defective nef, env, and vpr was constructed. The luciferase reporter gene was inserted into the nef gene sequence. The EBOV membrane GP protein expression plasmid was co-transfected with pNL 4-3.Luc.RE, and a pseudovirus containing the Ebola membrane protein could be packaged to detect the neutralizing antibody titer[20].
In this study, the expression plasmid pCAGGS-EBOV GP containing the full-length Zaire-type EBOV membrane protein gene was constructed and used to co-transfect human embryonic kidney 293 (HEK293) cells with pNL 4-3.Luc.RE plasmid. Collection of the cell supernatant and ultracentrifugation steps were used to prepare the pseudovirus pEBOV containing the GP protein. EBOV neutralizing antibody MIL 77 and control antibody confirmed that pEBOV can be used for the evaluation of neutralizing antibodies. Finally, EBOV neutralizing antibody detection was performed on the sera of the two vaccine-immunized mice by pEBOV.
-
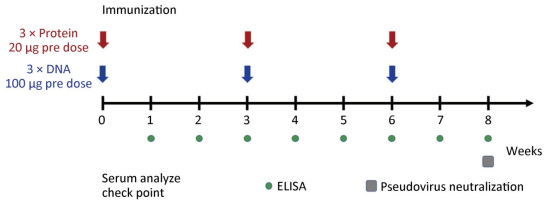
Female BALB/C mice were selected at 6-8 weeks for immunological experiments and were divided into recombinant protein group, DNA vaccine group, and control group, with five mice in each group. The protein group was immunized at 0, 3, and 6 weeks at a dose of 20 μg each time by mixing 20 μg recombinant protein in 50 μL PBS with an equal volume of QuickAntibody adjuvant (Boaolong Immunotechnology Co., Ltd.) followed by intramuscular injection of a leg. The pVR-GP-Fc DNA vaccine group was immunized at 0, 3, and 6 weeks at a dose of 100 μg each time. The immunization method used 100 μL of 1 mg/mL vaccine plasmid solution using the TERESA live gene importer (Shanghai Tarissa Biotechnology) delivered to leg muscle cells of the inoculated mice. The control group was injected with PBS or pVR empty vector. One to eight weeks after immunization of the mice, serum was collected by eyelid blood collection every week for indirect ELISA detection of binding titers. The sera collected at week 8 were mixed and used for neutralization test of pseudovirus (Figure 1).
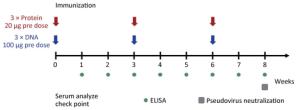
Figure 1. Strategy of immunization of mice with recombinant protein vaccine and DNA vaccine.
-
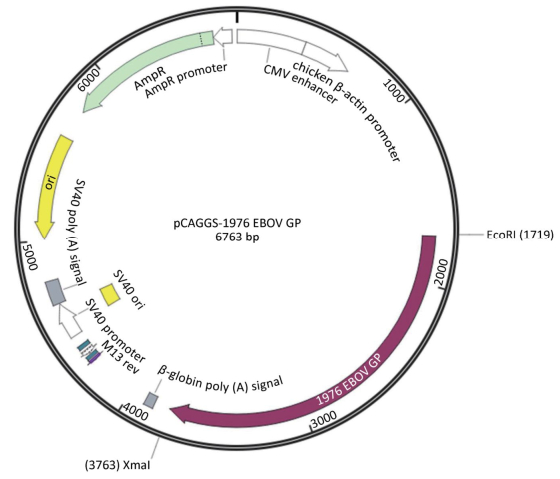
Zaire EBOV GP gene sequence (GenBank accession no. U23187) was selected, and an A (adenosine deoxynucleotide) site was added to the 1, 021 nucleic acid site in the U23187 sequence, so that the gene sequence could be read and translated to 676 amino acids. The EcoRI and Sam Ⅰ restriction sites were added to the 5' and 3' ends of the full-length GP protein sequence. The synthesis of the sequence was performed by Sino-Thai Biotech Co., Ltd. The synthesized sequence was double-digested with EcoRI and Sam Ⅰ into the pCAGGS vector, while the expression plasmid pCAGGS-EBOV GP was formed (Figure 2).
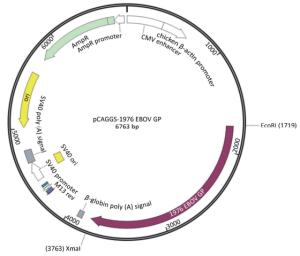
Figure 2. Plasmid map of pCAGGS-EBOV GP.
The EBOV GP protein expression plasmid pCAGGS-EBOVGP was transfected into HEK293T (Life Technologies, USA) cells, and the EBOV neutralizing antibody MIL-77 was used as the primary antibody for immunofluorescence detection to determine whether the expression plasmid was correctly expressed in the cells. HEK293T cells were seeded in 6-well plates at 8 × 105 cells/mL with 2 mL. After culture for 18-24 h, 3 μg of pCAGGS-EBOVGP plasmid and 9 μL of 1 mg/mL PEI (25 kD linear PEI, Polysciences, USA) were diluted in 50 μL of Opti-MEM medium. The DNA and PEI solution were mixed and kept at room temperature for 20 min[21]. The plasmid transfection mixture was added dropwise to a 6-well plate of HEK293T cells, and cultured in a 5% CO2 incubator at 37 ℃ for 48 h.
The cells transfected with the expression plasmid were removed by washing with PBS, and cold acetone was added to fix the cytospin slides (empty vector transfected cells as a control). PBS containing 10% fetal bovine serum (FBS) was used to block cells at 37 ℃ for 30 min. The cells were washed five times with PBS. The final suspension was added dropwise to PBS containing 50 μg/mL of MIL-77 and 10% FBS, and that suspension was dispensed dropwise on the cell surface for 1 h at 37 ℃. After washing the cells with PBS five times, fluorescein isothiocyanate- labeled goat anti-human IgG antibody (Zhongsu Jinqiao Company, Beijing) was diluted 1:50 in PBS containing 10% FBS and 0.01% Evans blue, and incubated on the cell surface at 37 ℃ for 1 h. The cells were then washed five times with PBS. Fluorescence was observed under an inverted fluorescence microscope using a 50% glycerol seal.
-
HEK293T cells in logarithmic growth phase were suspended in Dulbecco's modified Eagle's medium (DMEM) lacking penicillin-streptomycin (PS) and 10 mL of the cell suspension (9 × 105 cells/mL) was added to a 100 mm cell culture dish and cultured for 18-24 h. The cells were grown under surface growth was approximately 80% confluent. The micrograms of pNL 4-3.Luc.RE backbone plasmid and 8 μg of pCAGGS-EBOV GP membrane protein expression plasmid were diluted in 500 μL of Opti-MEM medium. Thirty-five microliters of 1 mg/mL PEI was diluted in the same manner and allowed to stand at room temperature for 3 min. Then the plasmid dilution and PEI were prepared. The dilutions were mixed and allowed to stand at room temperature for 20 min to form a transfection complex, which was pipetted into the culture dish with a micropipette, gently mixed, and placed in a thermostat. After 6 h of transfection, the cells were washed with PBS, and the medium was changed to DMEM containing 1% PS. Incubate in a 5% CO2 atmosphere at 37 ℃. Forty eight hours after transfection, the cell culture supernatant was collected, centrifuged at 3, 000 ×g for 20 min to remove cell debris, filtered through a 0.45-μm filter, and ultracentrifuged (Beckman, Germany) to concentrate the pseudovirus. Two-hundred microliters of concentrated pseudovirus per tube was stored at -80 ℃[22].
-
The successfully packaged pseudovirus pEBOV envelope contains a GP protein trimer and the virus particle contains a luciferase expression gene. After infecting sensitive cells, luciferase activity can be measured to determine the infection ability of the pseudovirus. The neutralizing antibody binds to the GP protein on the pseudovirus, which can block the pseudovirus infection. Using the neutralizing antibody MIL-77 as a positive control, a Mers-NP monoclonal antibody that does not bind to the Ebola GP protein was used as a negative control. After mixing with the pEBOV pseudovirus, Lenti-X 293T (ClonTech) cells were infected. Whether the pEBOV packaged successfully was determined by measuring the luciferase activity. The neutralizing antibody and the negative control antibody were diluted in DMEM-D medium (DMEM + 10% FBS + 25 mmol/L HEPES + 1% PS + 15 μg/mL DEAE-Dextran), and 25 μL per well was added to the 96-well plate at the same dilution. Three duplicate wells were used for each sample. The concentrated pEBOV was diluted 100 times in DMEM-D, then 25 μL was added to each sample well as a virus control. The mixture was placed in a 37 ℃ incubator, incubated for 1.5 h, and Lenti-X293T (ClonTech, USA) in good growth was digested. The cells were suspended in DMEM-D to a concentration of 6 × 105 cells/mL. Fifty microliters of the cell suspension was added to the incubated mixture, mixed, and placed in a 5% CO2 atmosphere at 37 ℃ for 48 h. The cultured cells were supplemented with 50 μL of LBright-Glo fluorescein reagent in each well, and allowed to stand for 3 min in the dark. One hundred microliters of the contents were aspirated from the 96-well plate to a fluorescence-detecting 96-well plate. A biocemimeter was used to determine the relative light unit (RLU) values.
-
Five days after the last immunization, blood samples were collected from the mice. Sera were separated and mixed within the same group at an equal ratio. The sera were inactivated at 56 ℃ for 30 min. The neutralizing antibody titers were detected by a micro-neutralization assay with the pseudotyped virus system. The titers of neutralizing antibodies were measured with the maximum dilution that achieving the median neutralization (ND50) determined.
-
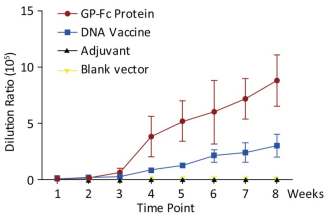
Both the purified recombinant protein GP-Fc and the gene vaccine pVR-GP-Fc containing the recombinant protein sequence induced antibodies against GP protein in mice. For the recombinant adjuvant, the recombinant protein induced the production of binding antibodies faster than the genetic vaccine, and the titer was higher. In the first week after immunization of the two vaccines, the endpoint of antibody titration in serum exceeded 103, and both vaccines rapidly induced specific immune production. There was no significant difference in antibody titer induced by the two vaccines 2 weeks after priming. After the third week of boosting, the recombinant protein induced IgG production was faster and stronger, and the serum dilution was 8.8 × 105 after two boosts. Specific antibodies could still be detected afterwards. The ability of the gene vaccine to induce antibody production was slightly worse than that of the recombinant protein. Moreover, at week 8 (i.e., two boosts) the antibody titer reached 3 × 105. Consistent with related studies, the results indicate that genetic vaccines induce humoral immune responses that are weaker than direct antigen immunity, both in strength and speed, compared to recombinant subunits (Figure 3).
Figure 3. Titer curves of binding valence each week after immunization.
-
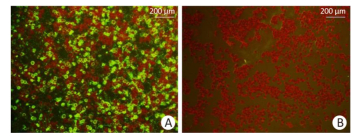
The immunofluorescence assay was carried out using the EBOV neutralizing antibody MIL-77 as the primary antibody. The cell surface of the transfected GP expression plasmid pCAGGS-EBOV GP showed obvious fluorescence signals, while the cells transfected with the empty vector did not produce any green signal (Figure 4). The constructed expression plasmid pCAGGS-EBOVGP can express EBOV GP protein in HEK293 cells with correct protein localization and molecular structure, and could stably bind to neutralizing antibodies. This plasmid could be used as a pseudovirus envelope plasmid.
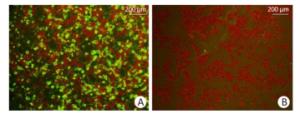
Figure 4. EBOV GP verification of the immunofluorescence result (×100); (A) HEK293 cells transfected with pCAGGS-EBOVGP; (B) HEK293 cells transfected with empty vector.
-
The micro-neutralization test of the prepared pseudovirus pEBOV and neutralizing antibody showed that it could be blocked by the EBOV neutralizing antibody MIL-77, while the Mers-NP monoclonal antibody could not block the virus infection (Table 1). RLU values were analyzed using GraphPad Prism software, t-test, with significant differences (t = 2.55, P = 0.0286 < 0.05).
Table 1. pEBOV Neutralize Verification Test
Concentration
(μg/mL)MIL-77
Neutralizing AntibodyMers-NP Monoclonal Antibody
Control AntibodyRLU Neutralization percentage (%) RLU Neutralization percentage (%) 20.00 3976.0 81.1 18039.0 14.3 5.00 7182.3 65.9 19116.3 9.2 1.25 10640.7 49.5 17941.0 14.8 0.31 13839.7 34.3 20025.7 4.9 0.08 17387.7 17.4 18523.7 12.0 The neutralization percentage regression equation y = 11.501 ln(x) + 47.038 was used to calculate the corresponding ND50 of the MIL-77 neutralizing antibody. The calculated ND50 was 1.29 μg/mL (Figure 5). The pEBOV prepared by this method could be used in the EBOV neutralization test.
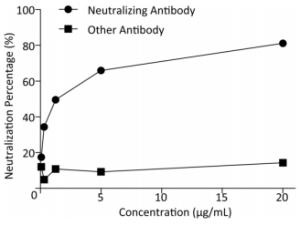
Figure 5. Variation of neutralizing percentage of MIL-77.
-
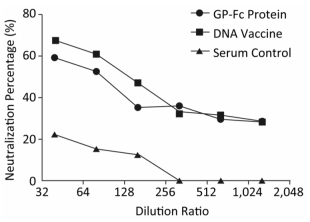
The identified Ebola pseudovirus pEBOV detection system was used to evaluate whether the recombinant protein GP-Fc and the genetic vaccine pVR-modGP-Fc could induce protective antibodies by a serum micro-neutralization test. The results of micro-neutralization experiments showed that both the recombinant protein GP-Fc and the gene vaccine pVR-modGP-Fc induced neutralizing antibodies in BALB/c mice. One-way ANOVA test analysis using GraphPad Prism software revealed a significant difference between the post-immune serum and the negative serum control (F = 13.29, P < 0.01). Both vaccine-induced neutralizing antibody half-inhibitor amounts (ND50) exceeded 1:80, and the percent neutralization at this dilution for the recombinant protein GP-Fc and the genetic vaccine pVR-modGP-Fc was 52.6% and 60.6%, respectively. Although the recombinant protein induced a better binding titer than the genetic vaccine, the neutralization test showed that the genetic vaccine pVR-modGP-Fc induced higher neutralizing titers in BALB/c mice. The test results are presented in Table 2, and the trend of the percentage of neutralization is shown in Figure 6.
Table 2. Neutralization Test of Vaccinate Mice Serum
Dilution Ratio GP-Fc Recombination Protein pVR-modGP-Fc DNA Vaccine Negative Serum Control RLU Neutralization percentage (%) RLU Neutralization percentage (%) RLU Neutralization percentage (%) 1:40 12953.0 59.5 10358.3 67.6 24736.7 22.6 1:80 15159.3 52.6 12597.3 60.6 27080.7 15.3 1:160 20677.7 35.3 16828.3 47.4 27977.7 12.5 1:320 20472.3 36.0 21543.7 32.6 37679.7 0.0 1:640 22451.0 29.8 21886.0 31.5 33855.0 0.0 1:1, 280 22784.3 28.7 22885.0 28.4 33328.7 0.0 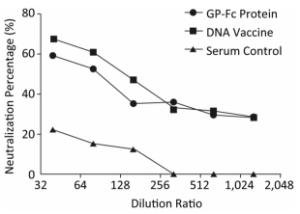
Figure 6. Serum neutralization test results
-
After the discovery of the EBOV, teams in the United States and Canada began to develop vaccines. In 1980, the U.S. team developed an inactivated virus vaccine that successfully passed the guinea pig challenge test, but subsequent experiments found that the protection of non-human primates was poor[6]. In 2005, the replication-defect EBOV was prepared by researchers of the University of Wisconsin. They replaced the VP30 gene of the virus and showed that the virus lost its ability to replicate. It could only proliferate in the cell line expressing VP30. Other features of the virus and wild strain were same[23]. In the animal infection immunization test, the replication-deficient virus was very safe and did not cause disease in mice with natural immunodeficiency. It also had good immunogenicity, and the protection conferred by induction was also good. The preparation passed the animal challenge test. However, due to the inherent inability to replicate in the body, protection requires a long time and repeat immunizations, and there is a risk of reorganization with wild plants in the high-incidence area, which may be has some security issues.
cAd3-EBOV was developed by the Institute of Allergy and Infectious Diseases of the National Institutes of Health[24]. The researchers integrated the Ebola GP protein sequence into the chimpanzee type 3 adenovirus vector, and used this vaccine to single-immunize 1010 viral particles of recombinant adenovirus. After 5 weeks, non-human primates were fully protected[7]. To improve the persistence of protection, the team introduced a prime-boost immunization strategy. After 8 weeks of cAd3-EBOV immunization, MVA (an Ebola vaccine based on the vaccinia virus) was obtained, which conferred good long-term protection[25]. The vaccine entered Phase Ⅲ clinical trials in 2015.
The National Microbiology Laboratory of the Public Health Agency of Canada developed the recombinant herpes stomatitis virus rVSV-ZEBOV vaccine in 2004[11]. The researchers replaced the G protein of VSV with the GP protein of EBOV. Compared with the adenovirus vector, the rVSV-ZEBOV surface molecule contains GP protein, which produces infectivity that is similar to the EBOV itself and can directly induce antibodies. The vaccine could simultaneously induce significant humoral and cellular immune responses and passed the animal challenge test[26-27]. Further research has found that it can also be used for emergency prevention and treatment after exposure to the virus. Rhesus monkeys were vaccinated within 30 min after contact with Zaire-type EBOV, with a protection rate of 50%[28-29]. The immunization dose required for rVSV-ZEBOV is not high, and 106 viral particles can achieve complete protection. The vaccine was put into emergency use in 2015 without a clinical Phase Ⅲ trial. The follow-up survey data released in 2016 showed that rVSV-ZEBOV was safe and effective, and the protective effect was good. Among the 5, 837 people who received the vaccine, 10 days or more after the inoculation Ebola cases were not recorded for a protracted time. In contrast, among those who did not receive the vaccine, 23 cases were recorded during the same period[30].
The recombinant protein vaccine and DNA vector vaccine constructed by our research group are more suitable for use in economically underdeveloped areas than the viral vector vaccines. Previous studies have also confirmed that both vaccines can stimulate high levels of antibodies, but the two vaccines can be protective only if they induce neutralizing antibodies. This study used a pseudovirus system based on the HIV sequence, which consists of the pNL 4-3.Luc.R-E backbone and an expression plasmid providing a membrane protein. The EBOV GP protein expression plasmid was used to replace the HIV membrane protein expression plasmid, and a pseudovirus with EBOV membrane protein as the core of HIV virus particles was constructed. The virus can mimic the process by which EBOV infects sensitive cells. At the same time, because of the luciferase-expressing gene, whether the virus is successfully infected can be determined by measuring luciferase activity. Using this system, we evaluated the neutralizing antibody titers of the recombinant protein GP-Fc and the genetic vaccine pVR-modGP-Fc. The neutralization titers of both exceeded 1:80. Moreover, the protective activity induced by the genetic vaccine was stronger than that of the recombinant protein GP-Fc. This was inconsistent with the results of the binding titer test. This result may be because the genetic vaccine allows the GP-Fc protein to be expressed directly by mouse cells with a structure similar to the Ebola GP protein of the viral protein. The change in pH and ion concentration during purification of the expressed protein in vitro could denature some of the proteins. In addition, the HEK293 system may be partially over-glycosylated. These factors result in a recombinant protein induced neutralizing antibody titer that is slightly lower than the DNA vector vaccine.
-
Thanks to Professor LU Ming from the Academy of Military Medical Sciences for providing MIL-77 neutralizing antibodies.
-
ZHANG Xiao Guang, and LI Xiao Jing conceived the idea and designed the study; YANG Ren, ZHU Ying, MA Jing, HAO Yan Zhe, WANG Xuan, HOU Mei Ling, and LIU Li Ping conducted all the experiments; YANG Ren analyzed the data. ZHANG Xiao Guang, YANG Ren, LI Xiao Jing, and FAN Li Yun, CAO Yu Xi wrote the manuscript and prepared all the tables and figures; ZHANG Xiao Guang reviewed the manuscript; all authors approved the manuscript.
doi: 10.3967/bes2018.097
Neutralizing Antibody Titer Test of Ebola Recombinant Protein Vaccine and Gene Vector Vaccine pVR-GP-FC
-
Abstract:
Objective In previous studies, we immunized mice with Ebola recombinant protein vaccine and gene vector vaccine. Both stimulated high levels of humoral immunity. In this work, we constructed a pseudovirus containing Ebola membrane proteins to verify whether the two immunization strategies can induce neutralizing antibodies in mice. Methods A pseudovirus containing an Ebola virus membrane protein based on the HIV-1 viral gene sequence was constructed and evaluated using a known neutralizing antibody. The titer of the neutralizing antibody in the sera of mice immunized with the recombinant protein and the gene vector vaccine was examined using a neutralization test. Results Ebola pseudovirus was successfully prepared and applied for neutralizing antibody detection. Immunological experiments showed that recombinant protein GP-Fc and gene vaccine pVR-modGP-Fc had good immunogenicity. The titer of the bound antibody in the serum after 8 weeks of immunization in mice was more than 1:105, and the recombinant protein induced greater humoral immunity. The results of the neutralization test based on the Ebola pseudovirus system demonstrated that both vaccines induced production of protective antibodies, while the gene vaccine induced a higher titer of neutralizing antibodies. Conclusion An Ebola pseudovirus detection system was successfully established and used to evaluate two Ebola vaccines. Both produced good immunogenicity. The findings lay the foundation for the development of new Ebola vaccines and screening for neutralizing monoclonal antibodies. -
Key words:
- Ebola virus /
- Recombinant subunit vaccine /
- DNA vaccine /
- Neutralizing antibody
-
Figure 1. Strategy of immunization of mice with recombinant protein vaccine and DNA vaccine.
Figure 4. EBOV GP verification of the immunofluorescence result (×100); (A) HEK293 cells transfected with pCAGGS-EBOVGP; (B) HEK293 cells transfected with empty vector.
Table 1. pEBOV Neutralize Verification Test
Concentration
(μg/mL)MIL-77
Neutralizing AntibodyMers-NP Monoclonal Antibody
Control AntibodyRLU Neutralization percentage (%) RLU Neutralization percentage (%) 20.00 3976.0 81.1 18039.0 14.3 5.00 7182.3 65.9 19116.3 9.2 1.25 10640.7 49.5 17941.0 14.8 0.31 13839.7 34.3 20025.7 4.9 0.08 17387.7 17.4 18523.7 12.0  下载: 导出CSV
下载: 导出CSV
Table 2. Neutralization Test of Vaccinate Mice Serum
Dilution Ratio GP-Fc Recombination Protein pVR-modGP-Fc DNA Vaccine Negative Serum Control RLU Neutralization percentage (%) RLU Neutralization percentage (%) RLU Neutralization percentage (%) 1:40 12953.0 59.5 10358.3 67.6 24736.7 22.6 1:80 15159.3 52.6 12597.3 60.6 27080.7 15.3 1:160 20677.7 35.3 16828.3 47.4 27977.7 12.5 1:320 20472.3 36.0 21543.7 32.6 37679.7 0.0 1:640 22451.0 29.8 21886.0 31.5 33855.0 0.0 1:1, 280 22784.3 28.7 22885.0 28.4 33328.7 0.0
下载: 导出CSV
-
[1] Johnson KM, Lange JV, Webb PA, et al. Isolation and partial characterisation of a new virus causing acute haemorrhagic fever in Zaire. Lancet, 1977; 1, 569-71. http://www.sciencedirect.com/science/article/pii/S0140673677920001 [2] Kuhn JH, Bao Y, Bavari S, et al. Virus nomenclature below the species level:a standardized nomenclature for laboratory animal-adapted strains and variants of viruses assigned to the family Filoviridae. Arch Virol, 2013; 158, 1425-32. doi: 10.1007/s00705-012-1594-2 [3] Lawrence P, Danet N, Reynard O, et al. Human transmission of Ebola virus. Curr Opin Virol, 2017; 22, 51-8. doi: 10.1016/j.coviro.2016.11.013 [4] Dye C. Ebola virus disease in West Africa——the first 9 months. N Engl J Med, 2015; 372, 188-9. doi: 10.1056/NEJMc1413884 [5] Kaner J, Schaack S. Understanding Ebola:the 2014 epidemic. Global Health, 2016; 12, 53. doi: 10.1186/s12992-016-0194-4 [6] Lupton HW, Lambert RD, Bumgardner DL, et al. Inactivated vaccine for Ebola virus efficacious in guineapig model. Lancet, 1980; 2, 1294-5. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=0520071204591347 [7] Kobinger GP, Feldmann H, Zhi Y, et al. Chimpanzee adenovirus vaccine protects against Zaire Ebola virus. Virology, 2006; 346, 394-401. doi: 10.1016/j.virol.2005.10.042 [8] Sullivan NJ, Martin JE, Graham BS, et al. Correlates of protective immunity for Ebola vaccines:implications for regulatory approval by the animal rule. Nat Rev Microbiol, 2009; 7, 393-400. doi: 10.1038/nrmicro2129 [9] Ye L, Yang C. Development of vaccines for prevention of Ebola virus infection. Microbes Infect, 2015; 17, 98-108. doi: 10.1016/j.micinf.2014.12.004 [10] Yang L, Li J, Gao GF, et al. Overview of Ebola virus vaccine. Sheng Wu Gong Cheng Xue Bao, 2015; 31, 1-23. http://d.old.wanfangdata.com.cn/Periodical/swgcxb201501001 [11] Garbutt M, Liebscher R, Wahl-Jensen V, et al. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J Virol, 2004; 78, 5458-65. doi: 10.1128/JVI.78.10.5458-5465.2004 [12] Volchkov VE, Volchkova VA, Slenczka W, et al. Release of viral glycoproteins during Ebola virus infection. Virology, 1998; 245, 110-9. doi: 10.1006/viro.1998.9143 [13] Dolnik O, Kolesnikova L, Becker S. Filoviruses:Interactions with the host cell. Cell Mol Life Sci, 2008; 65, 756-76. doi: 10.1007/s00018-007-7406-2 [14] Falasca L, Agrati C, Petrosillo N, et al. Molecular mechanisms of Ebola virus pathogenesis:focus on cell death. Cell Death Differ, 2015; 22, 1250-9. doi: 10.1038/cdd.2015.67 [15] Konduru K, Bradfute SB, Jacques J, et al. Ebola virus glycoprotein Fc fusion protein confers protection against lethal challenge in vaccinated mice. Vaccine, 2011; 29, 2968-77. doi: 10.1016/j.vaccine.2011.01.113 [16] Zhang X, Yang R, Wang J, et al. Eukaryotic Expression and Immunogenic Research of Recombination Ebola Virus Membrane Protein Gp-Fc. Chinese Journal of Virology, 2016; 32, 8-13. (In Chinese) [17] Yang R, Zhang X, Wang J, et al. Construction and Immunogenic Research of a Recombination Zaire Ebola Virus Lipoprotein GDP-FCC DNA Vaccine. Chinese Journal of Virology, 2017; 33, 186-90. (In Chinese) http://jtp.cnki.net/bilingual/detail/html/BDXB201702010 [18] Chen Q, Tang K, Zhang X, et al. Establishment of pseudovirus infection mouse models for in vivo pharmacodynamics evaluation of filovirus entry inhibitors. Acta Pharm Sin B, 2018; 8, 200-8. doi: 10.1016/j.apsb.2017.08.003 [19] Wang LL, Chen Q, Zhou LN, et al. Study of gonadal hormone drugs in blocking filovirus entry of cells in vitro. Acta Pharmaceutica Sinica, 2015; 50, 1545-50. (In Chinese) http://www.ncbi.nlm.nih.gov/pubmed/27169275 [20] He J, Choe S, Walker R, et al. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol, 1995; 69, 6705-11. http://www.ncbi.nlm.nih.gov/pubmed/7474080 [21] Xie Q, Xinyong G, Xianjin C, et al. PEI/DNA formation affects transient gene expression in suspension Chinese hamster ovary cells via a one-step transfection process. Cytotechnology, 2013; 65, 263-71. doi: 10.1007/s10616-012-9483-9 [22] Kutner RH, Zhang XY, Reiser J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc, 2009; 4, 495-505. doi: 10.1038/nprot.2009.22 [23] Hoenen T, Groseth A, de Kok-Mercado F, et al. Minigenomes, transcription and replication competent virus-like particles and beyond:reverse genetics systems for filoviruses and other negative stranded hemorrhagic fever viruses. Antiviral Res, 2011; 91, 195-208. doi: 10.1016/j.antiviral.2011.06.003 [24] Sullivan NJ, Geisbert TW, Geisbert JB, et al. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature, 2003; 424, 681-4. doi: 10.1038/nature01876 [25] Stanley DA, Honko AN, Asiedu C, et al. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat Med, 2014; 20, 1126-9. doi: 10.1038/nm.3702 [26] Jones SM, Feldmann H, Stroher U, et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med, 2005; 11, 786-90. doi: 10.1038/nm1258 [27] Henao-Restrepo AM, Longini IM, Egger M, et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein:interim results from the Guinea ring vaccination cluster-randomised trial. Lancet, 2015; 386, 857-66. doi: 10.1016/S0140-6736(15)61117-5 [28] Feldmann H, Jones SM, Daddario-DiCaprio KM, et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog, 2007; 3, e2. doi: 10.1371/journal.ppat.0030002 [29] Geisbert TW, Daddario-DiCaprio KM, Williams KJ, et al. Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J Virol, 2008; 82, 5664-8. doi: 10.1128/JVI.00456-08 [30] Henao-Restrepo AM, Camacho A, Longini IM, et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease:final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet, 2017; 389, 505-18. doi: 10.1016/S0140-6736(16)32621-6 -

 点击查看大图
点击查看大图
图(6) / 表ll (2)
计量
- 文章访问数: 3735
- HTML全文浏览量: 954
- PDF下载量: 86
- 被引次数: 0

 Quick Links
Quick Links