

 下载:
下载:



-
Myoepithelioma (ME) is a rare tumor composed of myoepithelial cells that usually surround glandular tissue. MEs differ from mixed tumors as they present minimal or no ductal differentiation[1]. MEs can originate from soft tissue and bones, but histogenesis remains uncertain.
MEs seldom occur in the mediastinum; most cases described have reported that the tumor originated from ectopic salivary gland tissue along the bronchial mucous glands or tracheobronchial tree, and the mediastinum was secondarily involved. Only four well-documented cases have been reported in the English literature on MEs originally occurring in the mediastinum (Supplementary Table S1 available in www.besjournal.com). MEs are primarily found in the adult population but have also been described in children.
Table S1. Review of cases of primary myoepithelioma in mediastinum
Case Age/
sexSite Origin IHC profile Genetic
alterationBehavior Invasive growth Treatment/
outcomeCase 1[1] 24/M Left mediastinum Undefined
(Stem cells)CKAE1/AE3(+), CK7(+), Vimentin(+),
MIC2(CD99+), S100(+),
α-SMA(+),
p63(+), GFAP(+),
CD117 (F+),
WT1 (cytoplasmic)(F+)NA Benign No Resection/
NED, 6 moCase 2[2] 51/F Right posterior
mediastinumUndefined EMA(+), SMA(+), S-100(+)
CD99(+), calponin(F+), p63(F+),
GFAP (F+)EWSR1
rearrangementBenign No Resection/
NED, 24 moCase 3[3] 51/F Inferior top
mediastinumAtrial epicardium
or myocardiumCKAE1/AE3(+),
CK7(+), Vimentin(+),
S-100(+), Calretinin(+),
CEA(+)NA Benign Yes Resection/
NED, 7 moCase 4[4] 62/F Left posterior
mediastinumNA S100 (+), vimentin(+), EMA(F+),
CK8/18(F+)EWSR1
rearrangementBenign No Resection/
NACase 5
[The present]9/M Right posterior
mediastinumIntervertebral
foramina
(Stem cells)S-100(+), Desmin(+),
CK(+), CK19(+),
INI1(+), SMA(F+),
CD99 (F+)EWSR1
rearrangementBenign Yes Resection/
REC, 6 moNote. Abbreviations: M, male; F, female; NA, not available; IHC, immunohistochemistry; CK, cytokeratin; GFAP, glial fibrillary acid protein; F+, focally positive; SMA, alpha-smooth muscle actin; EMA, epithelial membrane antigen; WT, wilm tumor; NED, no evidence of disease; REC, recurrence; EWSR1, Ewing sarcoma breakpoint region1; CEA, carcinoembryonic antigen; mo, month. Here, we report a case of soft tissue ME in the mediastinum of a 9-year-old boy. A large heterogeneously enhanced soft tissue mass with multiple amorphous calcifications adjacent to the spine was detected in the right posterior mediastinum. The mass measured 7.3 × 5.9 × 7.4 cm3 and had infiltrated the tenth posterior rib (Figure 1). Immunohistochemical staining of the initial ultrasound-guided needle biopsy revealed positive expression for S-100, ERG, CD99 (locally), and Ki67 (3%) (Figure 2). Due to the invasiveness of the tumor, mesenchymal chondrosarcoma was the favored diagnosis, and one cycle of ifosfamide and doxorubicin was administered while the diagnosis was confirmed.
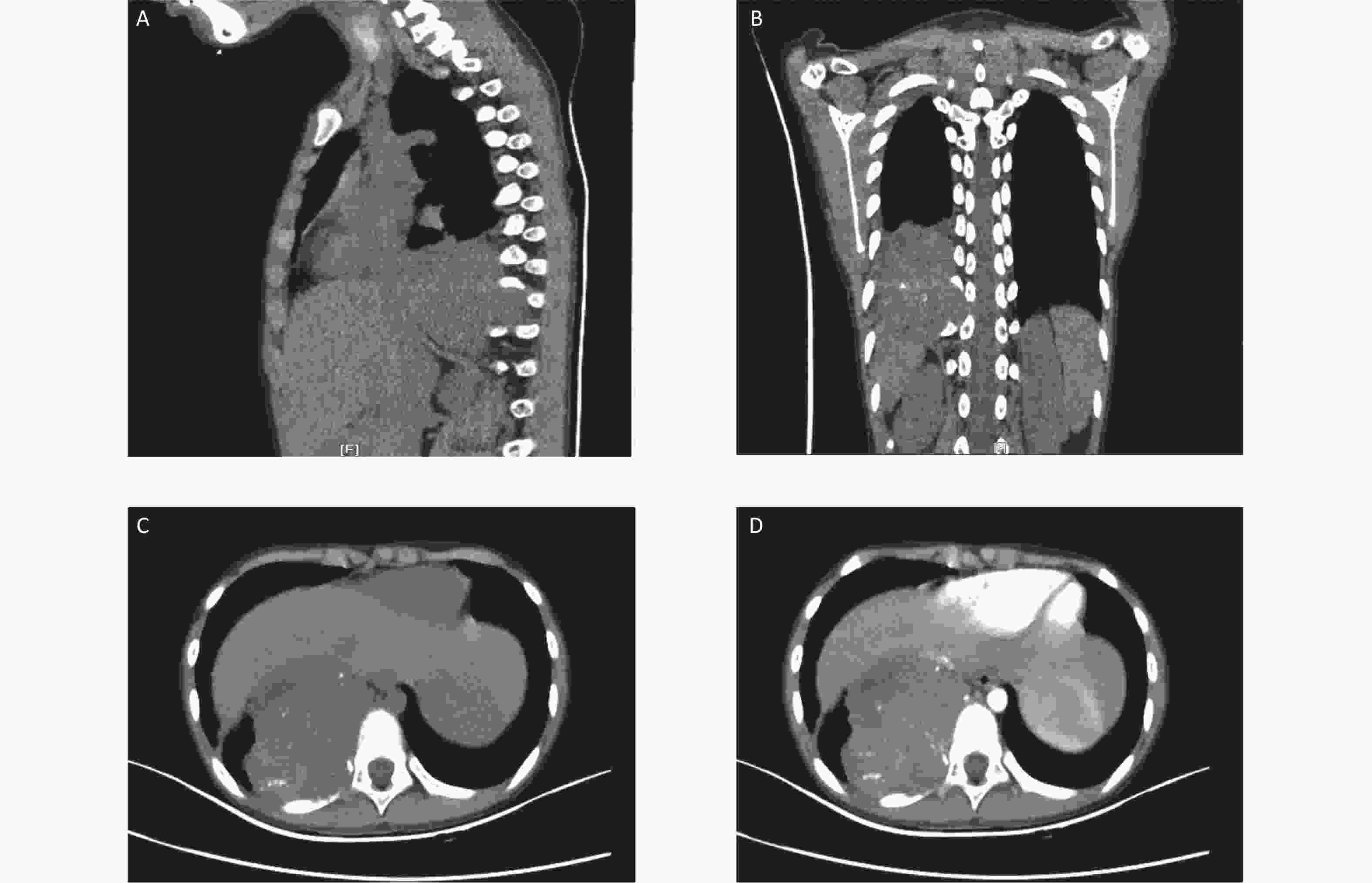
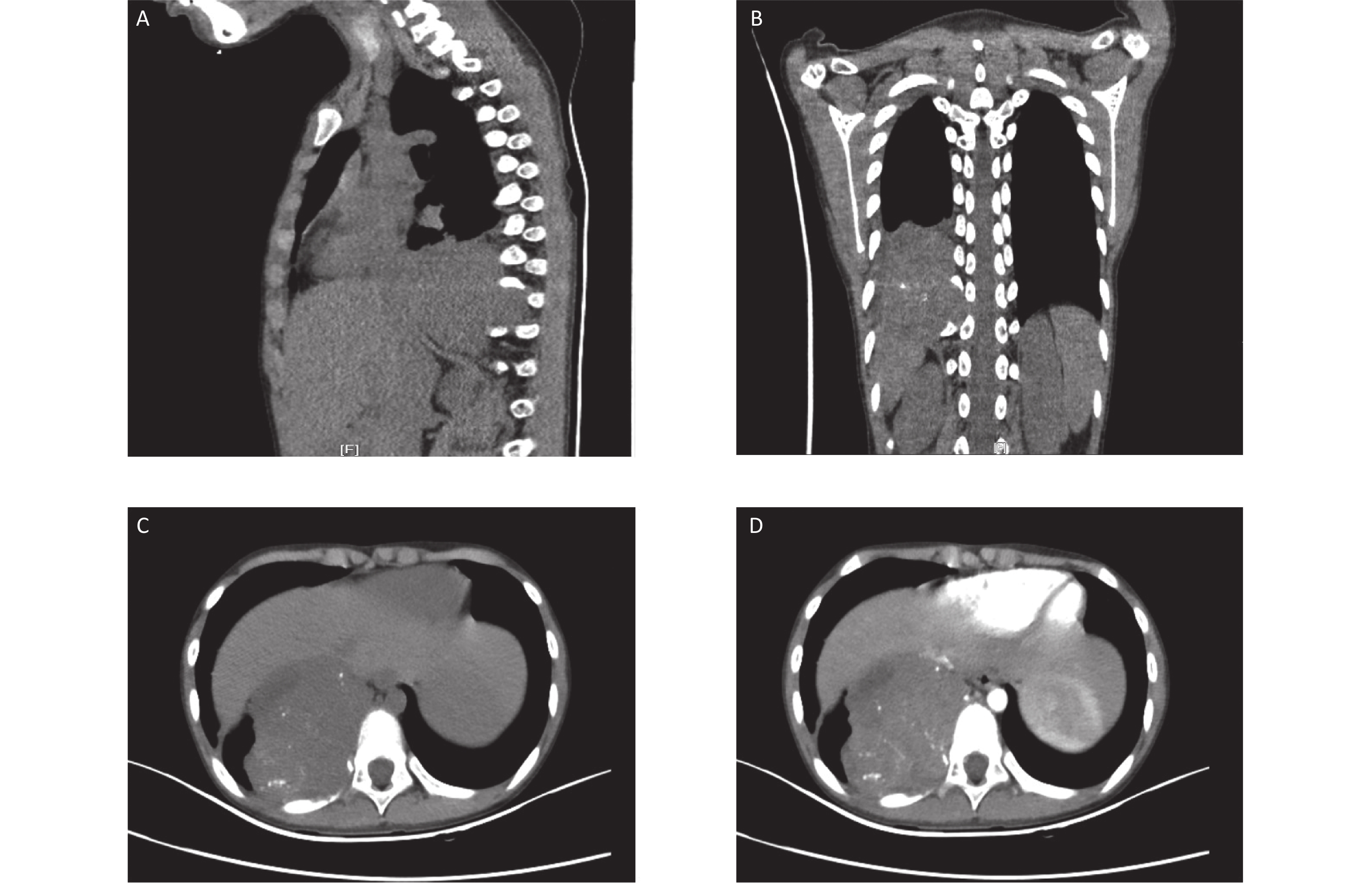
Figure 1. Computed tomography (CT) images at the initial diagnosis. The three-dimensional CT scan showing a mixed density tumor expanding to the intervertebral foramina (A and B). Contrast-enhanced CT showing a large heterogeneously enhanced soft tissue mass with multiple amorphous calcifications in the right posterior mediastinum infiltrating the tenth posterior rib (C and D).
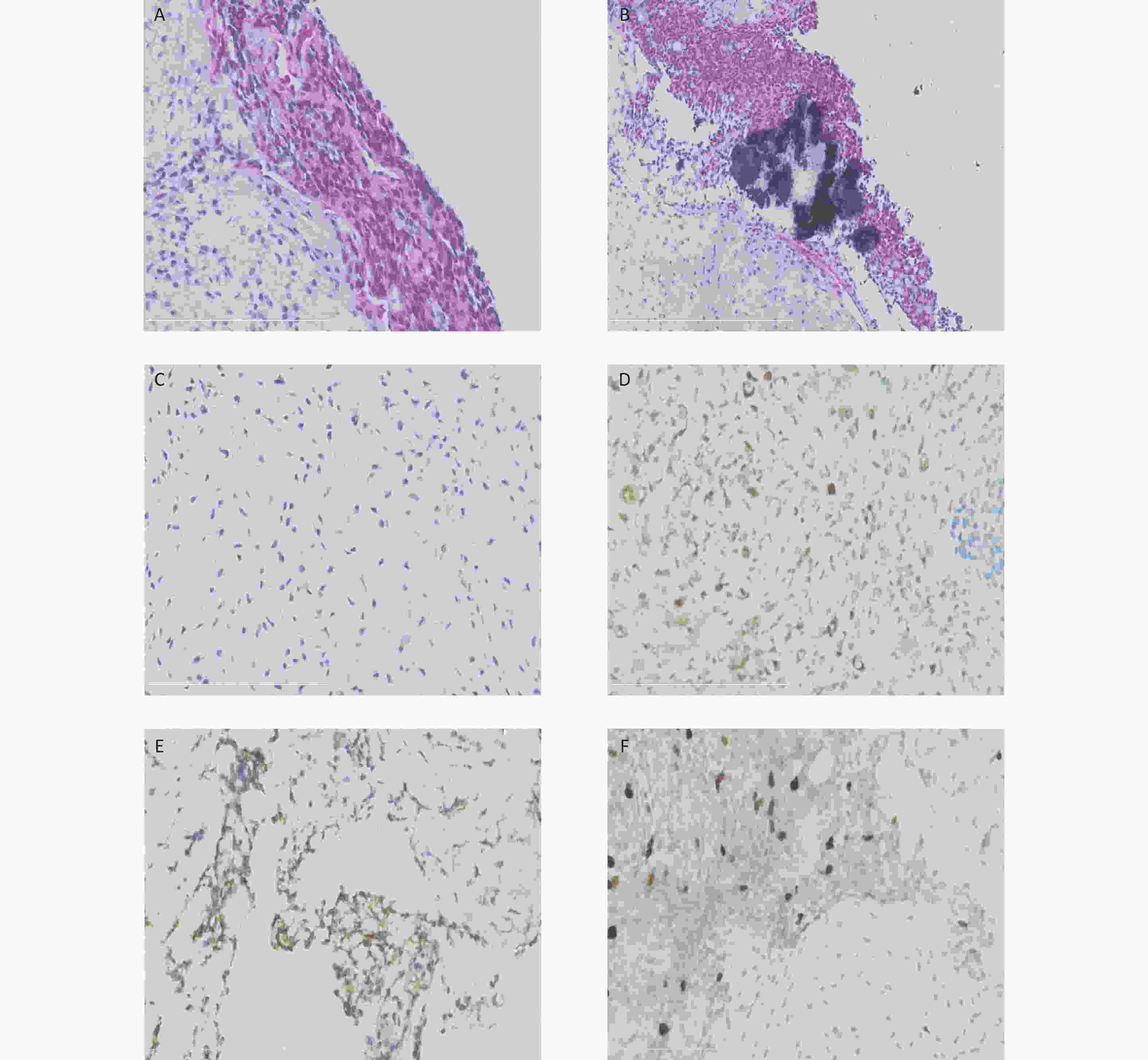
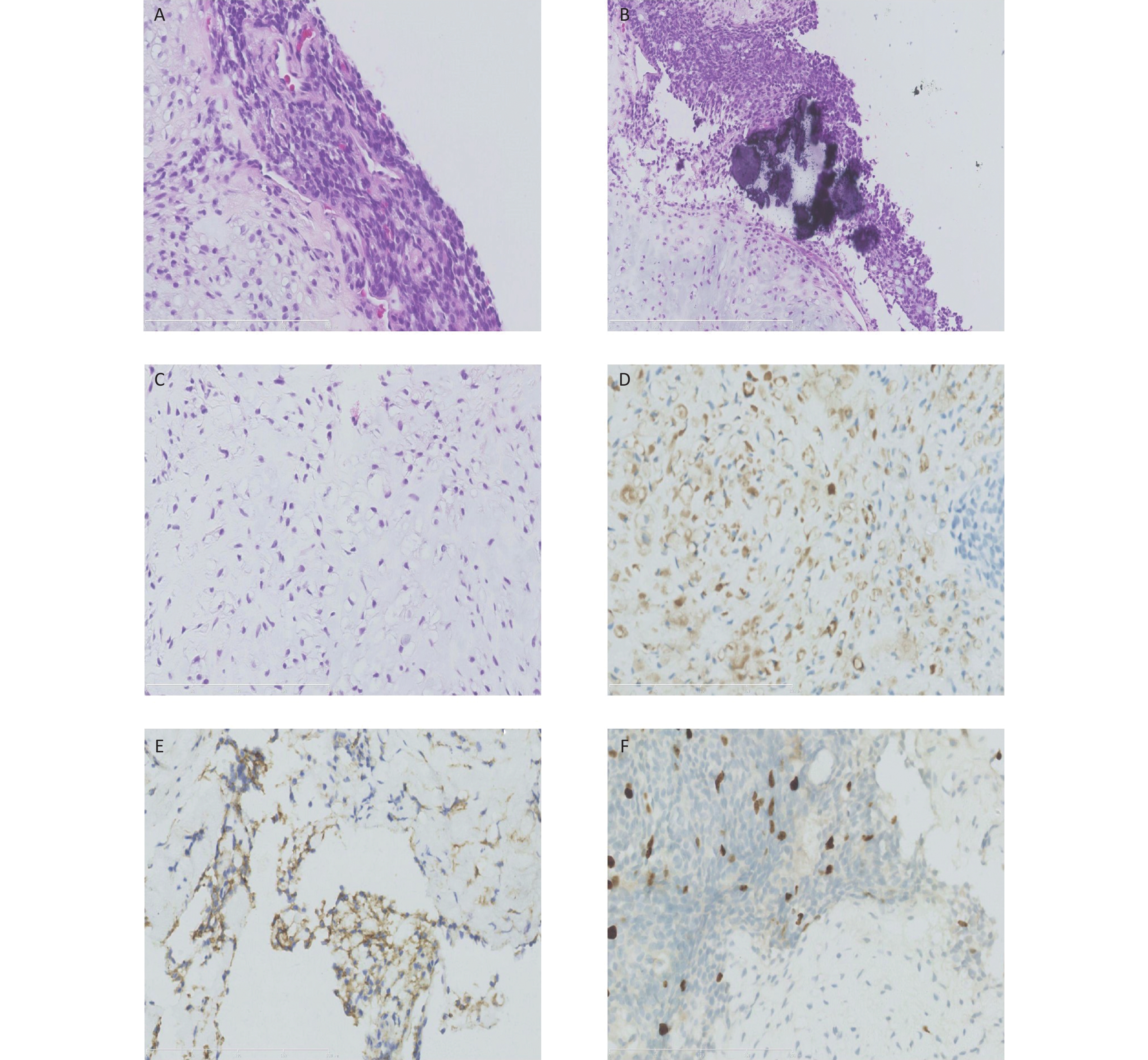
Figure 2. Histological and immunohistochemical results of the ultrasound-guided needle biopsies. Microscopic examination shows the tumor cells arranged in cords and sheets without ductal structures (A). The background was locally myxoid stroma with local calcification (B) and chondrometaplasia (C). Immunohistochemical staining reveals the positive expression of S-100 (D), CD99 (locally) (E), and Ki67 (3%) (F).
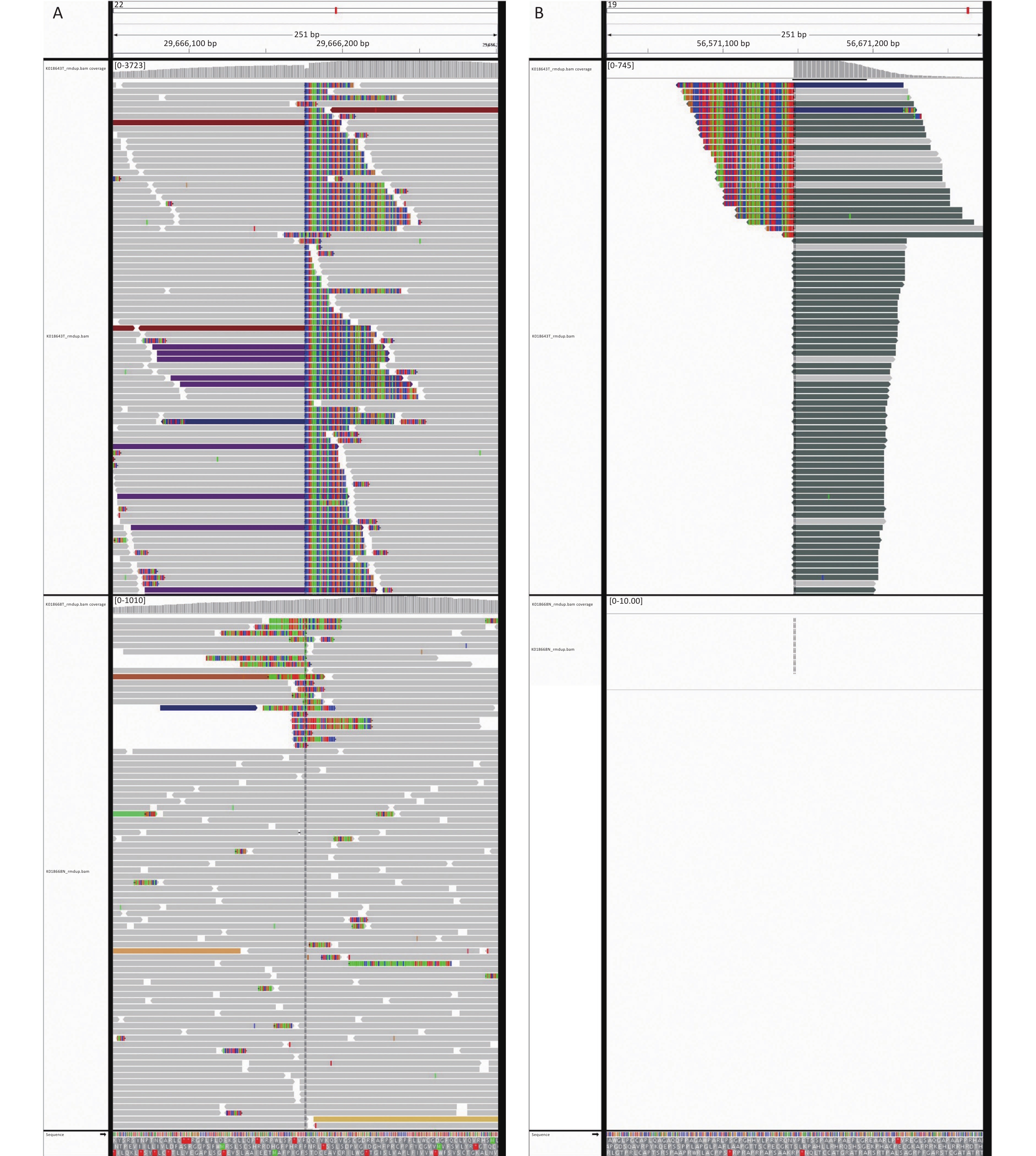
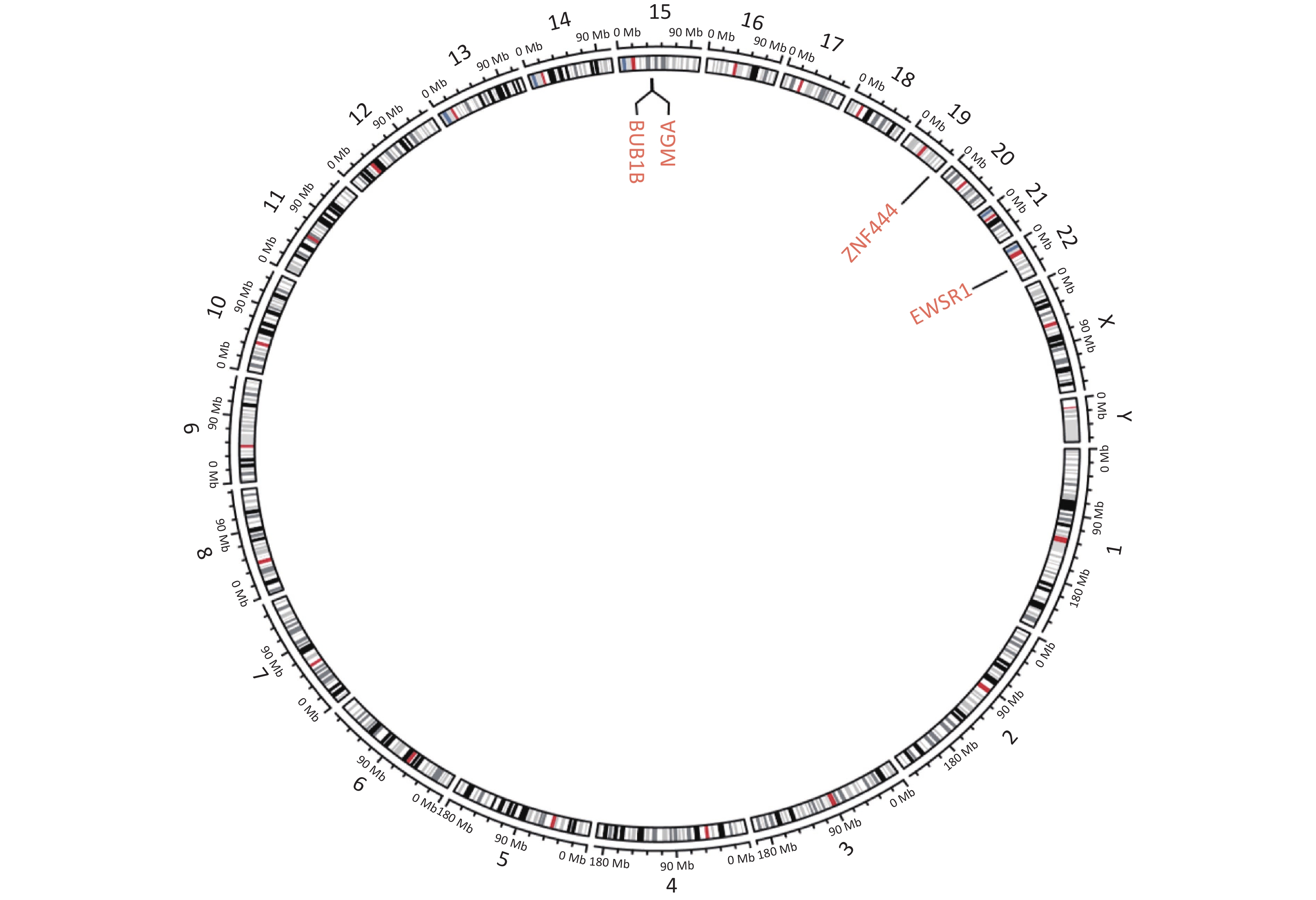
Next-generation sequencing (NGS) was performed considering the discrepancy between the proliferation index (Ki67, 3%) and the invasive growth. NGS revealed a EWSR1-ZNF444 gene fusion [t (19; 22) (q13; q12)], where exon 7 of EWSR1 (rearrangement position: 29686175 on chromosome 22) was fused in-frame to exon 5 of ZNF444 (rearrangement position: 56671149 on chromosome 19) (Figure 3, Supplementary Figure S1 available in www.besjournal.com). In 2008, Brandal et al. [2] first reported that the translocation of t (1; 22) (q23; q12) resulted in the fusion of EWSR1-PBX1 in soft tissue ME. To date, EWSR1 gene rearrangements have been detected in nearly half of soft tissue myoepithelial tumor cases, with several reported fusion partners, including PBX1 (1q23), ZNF444 (19q23), POU5F1 (6p21), ATF1 (12q13), and PBX3 (9q339). Therefore, the EWSR1-ZNF444 gene fusion suggested a diagnosis of ME. In addition to EWSR1 gene rearrangements, uncommon fusions of PLAG1, FUS, SRF, and OGT with fusion partners, such as FOXO3, have been reported in soft tissue ME. SMARCB1/INI1 deficiency is found in a significant subset of ME cases, particularly in malignant lesions [3]. Different types of gene fusions/mutations dominate in different MEs (Supplementary Table S2 available in www.besjournal.com). In addition, the present case carried a BUBIB germline mutation (Supplementary Figure S1). BUBIB encodes a key protein in the mitotic spindle checkpoint; however, its association with ME is unknown.
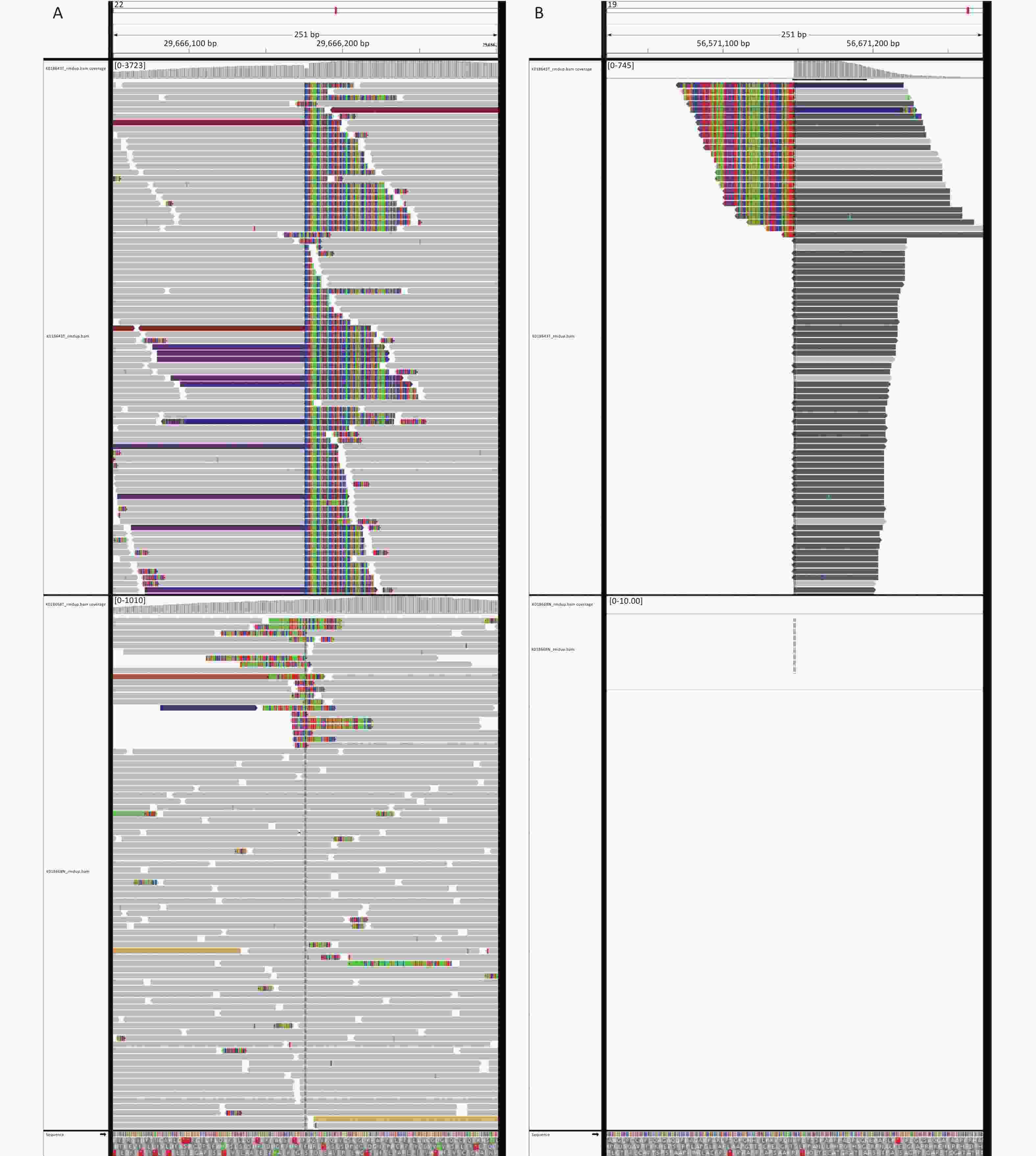
Figure 3. Next-generation sequencing results of the ultrasound-guided needle biopsies. Sarcoma PanScan analysis revealed an EWSR1-ZNF444 gene fusion, where exon 7 of EWSR1 (rearrangement position: 29686175 on chromosome 22) was fused in-frame to exon 5 of ZNF444 (rearrangement position: 56671149 on chromosome 19) (A and B).
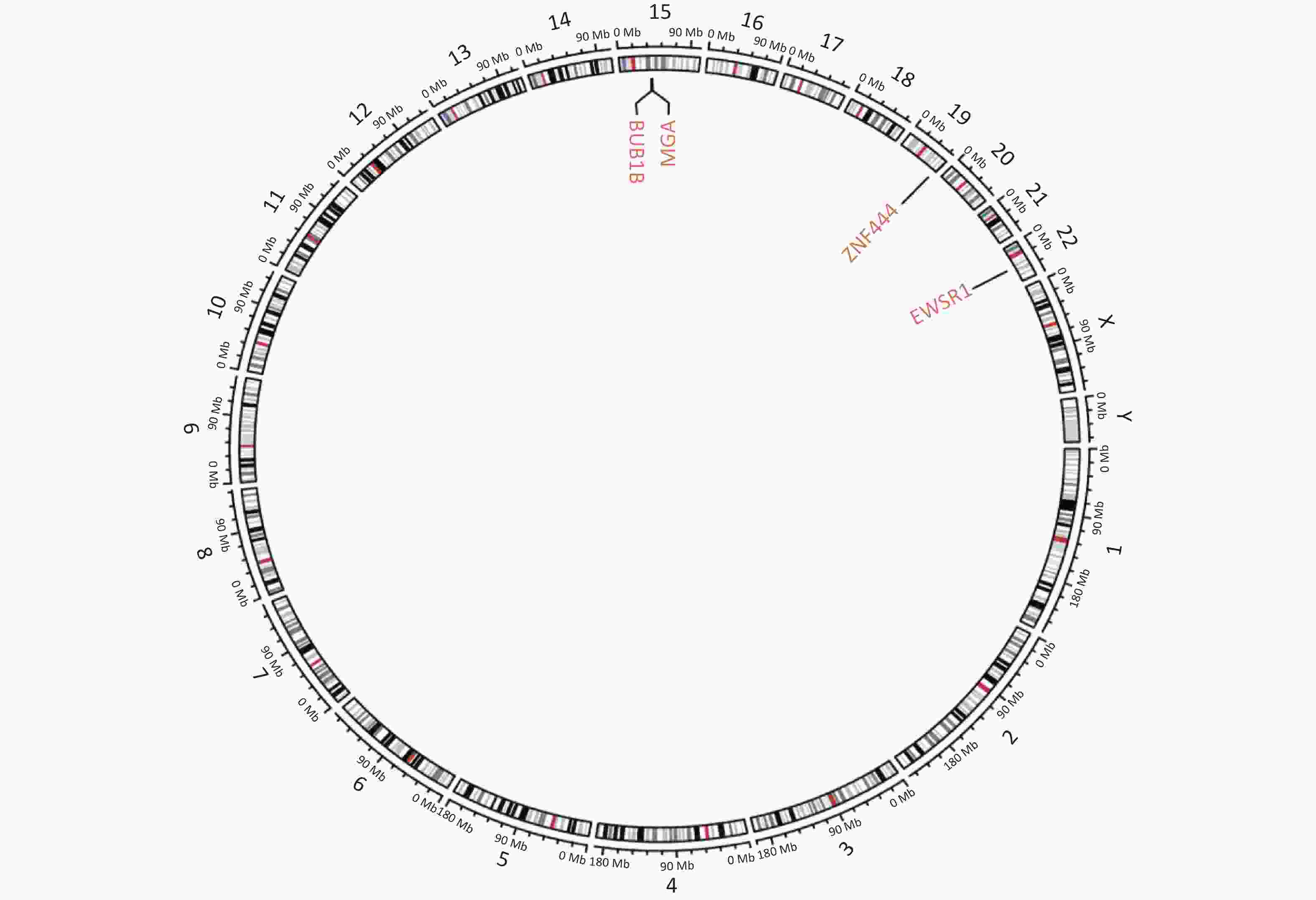
Figure S1. Circular genetic map of the result of next-generation sequencing. Circular genetic map revealed a Somatic MGA:c.6565 deletion, a Germline BUB1B:c.948-966 deletion and a Fusion EWSR1(E7): ZNF444(E5).
Table S2. Gene alteration in myoepithelioma of different sites
Tumor site Gene rearrangement Gene deficiency EWSR1 FUS OGT SRF PLAG1 RREB1 SMARCB1/ INI1 Soft tissue Yes[5] Yes[5] Yes[4, 6] Yes[7] Yes[8] Yes[9] Yes[10, 11] Bone Yes[5] Yes[12] NR NR NR NR NR Salivary gland No[5] NR NR NR Yes[13] NR NR Visceral locations (lung) Yes[5] Yes[5, 14] NR NR NR NR NR Skin Yes[15] NR NR NR Yes[16] NR NR MELTVR No[17] No[17] NR NR NR NR Yes[17] Note. Abbreviations: NR, not reported; MELTVR, myoepitheliom like tumors of the vulvar region; EWSR1, Ewing sarcoma breakpoint region1; FUS, fused in sarcoma gene; PLAG1, pleomorphic adenoma gene 1; OGT, O-linked N-acetylglucosamine; SRF, serum response factor; SMARCB, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1. Combined with the NGS results, a pathological consultation at an external institution favored ME; thus, thoracotomy was performed, and the tumor was excised in the hospital. The tumor originated from the 9–11 intervertebral foramina, and the local ribs were deformed and damaged. As myoepithelial cells were situated along the outer aspect of the acini, the intercalated ducts, and the interlobar and interlobular striated ducts, we assumed that the ME originated from stem cells in the intervertebral foramina capable of differentiation following the soft tissue ME classification. Soft tissue MEs are usually described in the subcutis and deep soft tissue (intramuscular, within the fascia, and subfascial). Our case is the first arising from the intervertebral foramina to be reported in the English language literature.
A microscopic examination of the postoperative mass revealed tumor cells arranged in cords and sheets with a rare adenoid arrangement and no ductal structure (data not shown). The tumor cells demonstrated mild atypia without evident nucleoli or mitoses. No tumors were found in the involved lung tissue. The diagnosis of benign ME was confirmed based on these morphological features. This was different from the malignant diagnosis provided by the initial ultrasound-guided needle biopsy. The tumor, in our case, had invaded the tenth posterior rib, which has never been reported. The criteria for malignant ME are different, but an invasive growth pattern is a useful criterion for establishing malignancy in a patient with salivary ME. However, Hornick and Fletcher [4] reported that this criterion does not apply to soft tissue myoepithelial tumors, as benign tumors with microscopically infiltrative margins do not recur or metastasize. Thus, they proposed that the appearance of moderate or severe cellular atypia was associated with malignancy in soft tissue myoepithelial tumors; border infiltration did not correlate with aggressive behavior, nor did the mitotic rate, as no specific mitotic rate cutoff exists [4]. Another study showed that a high Ki-67 labeling index (Ki67 LI, 14%–60%) may support a malignant diagnosis, as high Ki-67 LI is seen in severe nuclear atypia, and cases with mild to moderate nuclear atypia also exhibit a Ki-67 LI > 10% [5]. In the present case, the tumor cells were mildly atypic without an evident nucleolus or mitoses and a Ki67 concentration of 8% in postoperative tissue. Therefore, benign ME was finally diagnosed.
Adjuvant therapy was not administered in this case. Surgery has been the predominant treatment modality for soft tissue ME. As incomplete excision of benign MEs has been reported to lead to recurrence or metastasis, complete resection is recommended. A conservative excision with a minimum 5 mm margin of uninvolved tissue is supported for cases of benign ME with possible capsular/adjacent tissue infiltration [6]. Given its scarcity, the effects of adjuvant therapy on localized and metastatic diseases have not been clarified. Specialists support perioperative radiotherapy for malignant ME, where wide excision or amputation is suggested; chemotherapy has no distinct benefit. The prognosis for soft tissue ME is based on the histological appearance following the diagnostic criteria for malignant myoepithelial tumors. Hornick and Fletcher [4] studied 101 cases of myoepithelial tumors with moderate to severe cytological atypia and reported a local recurrence rate of 42% and a metastatic rate of 32%, whereas cytologically bland tumors recurred locally in 18% of cases and none metastasized. Another study showed that local relapse is seen in all tumor grades, with 5-year relapse rates of 29% vs. 64% for low-grade vs. high-grade tumors, respectively [7]. Our case recurred locally 6 months after surgery, which was attributed to the difficulty in completely removing the tumor from the intervertebral foramina.
Soft tissue ME occurs over a wide age range, with approximately 20% of the cases reported in children [8]. The biological behavior of pediatric ME is similar to that seen in adults, although soft tissue ME in children has a high rate of malignancy [8]. About 27 cases of soft tissue ME have been reported in patients < 18 years old (Supplementary Table S3 available in www.besjournal.com). Ectomesenchymal chondromyxoid tumor of the tongue, which has been classified into the pathological spectrum of soft tissue ME by the WHO, was not included. The 29 cases of soft tissue myoepithelial carcinoma in children reported by Gleason et al. [9] and the 7 cases reported by Bisogno et al. [10] were not included in our table because we could not distinguish which cases were mixed tumors. In total, 17 of 27 (63%) pediatric cases (including the present case) located in different anatomic regions were malignant; 3 of 9 cases of benign MEs demonstrated invasive growth, and 2 recurred.
Table S3. Review of soft tissue myoepithelioma in children below 18 years olda
Authors No. of
casesAge/
sexSite Gene
rearrangementBehavior Invasive
growthRecurrence Metastasis Treatment/
outcomeKilpatrick et al, 1997[18] 1 9/M axilla NA Malignant* Yes Yes No Resection/
REC, 30 moWaldrop et al, 2001[19] 1 11/M thigh NA Malignant Yes No Orbir, 10 mo Resection/
died 4 mo after
metastasisVan den Berg
et al, 2004[20]1 17/M left lower leg NA Malignant NA − LN, L, bone, 2 mo amputationb/
died 7 mo after
amputationHarada et al,
2005[21]1 17/M forearm NA Malignant Yes No L, 6 mo Resection/
died 18 mo
after metastasisHallor et al,
2008[22]1 11/F back NA Benign Yes Yes, 6 y L, 7 y Resection/
AwD,9 yHerlihy et al,
2009[23]1 3mo/M orbit NA Benign No No No Resection/
NED, 36 moGuedes et al,
2009[24]1 6/F forearm NA Malignant No No L, 1 mo Resection+
chemo/, NAAntonescu et al, 2010[5] 7 9/M arm EWSR1 Benign NA NA NA NA 7/F hip EWSR1 Malignant NA NA NA NA 11/F forarm EWSR1 Malignant NA NA NA NA 13/F peri-ocular EWSR1 Malignant NA NA NA NA 7/F peri-orbital EWSR1 Malignant NA NA NA NA 1/M head&neck EWSR1 Malignant NA NA NA NA 2/F mediastinum EWSR1 Malignant NA NA NA NA Rekhi et al,
2012[25]1 18/M inguinal No Benign Yes NA NA NA Antonescu et al, 2013[17] 3 3mo/M knee No Malignant NA NA NA NA 3/M mediastinum No Malignant NA NA NA NA 8/F thigh No Malignant NA NA NA NA Park et al,
2013[26]1 18/M forearm NA Benign No No No Resection/
NED, 4 yGupta et al,
2016[27]1 2/F CNS NA Malignant Yes Yes Cervical
spinechemo+Resection+XRT/AwD, 30 mo Baldovini et al, 2018[28] 1 newborn neck NA Malignant No No No Resection+chemo+XRT/NED, 12 mo Bhanvadia et al, 2017[29] 1 14/M gluteal NA Benign No No No Resection/
NADe Cates et al, 2020[30] 1 15/M skull base No Benign No No No Resection/
NED, 24 moSegawa et al,
2020[8]2 16/M groin EWSR1 Benign No No No Resection/
NED, 24 mo5/F back EWSR1 Benign No No No Resection/
NED, 3 moTrevinoa et al, 2020[10] 1 12/M foot
plantarNA Malignant No NA L, pericardium, RP Resection/
NAThe present
case1 9/M mediastinum EWSR1 Benign Yes Yes, 6 mo No Resection/
AwD, 7 moNote. *Mitoses numbered eight per ten. aCases with ductal differentiation/structures were excluded. bTNF-α, melphalan, perfusion followed by amputation. Abbreviations: chemo, chemotherapy; NA, not available; NED, no evidence of disease; XRT, radiation therapy; AwD, alive with disease; LN, lymph node; L, lung; RP, retroperitoneum; REC, recurrence; CNS, central nervous system; mo, month; y, year. Malignant ME predominance has been reported in children [8], and malignant MEs have a worse prognosis, with a mortality rate of 43% (after 26 months) in children vs. 13% (after 50 months) in adults [4, 9]. The location of the tumor and infiltration of the rib were discomforting features in our case. The recurrence after the surgery posed a difficult situation for treatment. Radiotherapy may harm the nerves passing through the intervertebral foramina, chemotherapy has no demonstrated distinct benefit in the literature, and repeated surgery can be traumatic for children. It is also unknown whether the lesion becomes malignant during recurrence. Therefore, more studies on the tumorigenesis mechanisms of ME are warranted.
Ethical Approval All procedures performed in studies involving human participants followed the ethical standards of the institutional and/or national research committee and the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. The committees of Shengjing Hospital of China Medical University approved the case details for publishing (approval number 2021PS619K).
Consent for Publication The patient’s parents provided written informed consent for the case details to be published.
doi: 10.3967/bes2022.112
Soft Tissue Myoepithelioma in Children: A Rare Case Report of the Mediastinum and a Review of the Literature
-
-
Figure 1. Computed tomography (CT) images at the initial diagnosis. The three-dimensional CT scan showing a mixed density tumor expanding to the intervertebral foramina (A and B). Contrast-enhanced CT showing a large heterogeneously enhanced soft tissue mass with multiple amorphous calcifications in the right posterior mediastinum infiltrating the tenth posterior rib (C and D).
Figure 2. Histological and immunohistochemical results of the ultrasound-guided needle biopsies. Microscopic examination shows the tumor cells arranged in cords and sheets without ductal structures (A). The background was locally myxoid stroma with local calcification (B) and chondrometaplasia (C). Immunohistochemical staining reveals the positive expression of S-100 (D), CD99 (locally) (E), and Ki67 (3%) (F).
Figure 3. Next-generation sequencing results of the ultrasound-guided needle biopsies. Sarcoma PanScan analysis revealed an EWSR1-ZNF444 gene fusion, where exon 7 of EWSR1 (rearrangement position: 29686175 on chromosome 22) was fused in-frame to exon 5 of ZNF444 (rearrangement position: 56671149 on chromosome 19) (A and B).
S1. Circular genetic map of the result of next-generation sequencing. Circular genetic map revealed a Somatic MGA:c.6565 deletion, a Germline BUB1B:c.948-966 deletion and a Fusion EWSR1(E7): ZNF444(E5).
S1. Review of cases of primary myoepithelioma in mediastinum
Case Age/
sexSite Origin IHC profile Genetic
alterationBehavior Invasive growth Treatment/
outcomeCase 1[1] 24/M Left mediastinum Undefined
(Stem cells)CKAE1/AE3(+), CK7(+), Vimentin(+),
MIC2(CD99+), S100(+),
α-SMA(+),
p63(+), GFAP(+),
CD117 (F+),
WT1 (cytoplasmic)(F+)NA Benign No Resection/
NED, 6 moCase 2[2] 51/F Right posterior
mediastinumUndefined EMA(+), SMA(+), S-100(+)
CD99(+), calponin(F+), p63(F+),
GFAP (F+)EWSR1
rearrangementBenign No Resection/
NED, 24 moCase 3[3] 51/F Inferior top
mediastinumAtrial epicardium
or myocardiumCKAE1/AE3(+),
CK7(+), Vimentin(+),
S-100(+), Calretinin(+),
CEA(+)NA Benign Yes Resection/
NED, 7 moCase 4[4] 62/F Left posterior
mediastinumNA S100 (+), vimentin(+), EMA(F+),
CK8/18(F+)EWSR1
rearrangementBenign No Resection/
NACase 5
[The present]9/M Right posterior
mediastinumIntervertebral
foramina
(Stem cells)S-100(+), Desmin(+),
CK(+), CK19(+),
INI1(+), SMA(F+),
CD99 (F+)EWSR1
rearrangementBenign Yes Resection/
REC, 6 moNote. Abbreviations: M, male; F, female; NA, not available; IHC, immunohistochemistry; CK, cytokeratin; GFAP, glial fibrillary acid protein; F+, focally positive; SMA, alpha-smooth muscle actin; EMA, epithelial membrane antigen; WT, wilm tumor; NED, no evidence of disease; REC, recurrence; EWSR1, Ewing sarcoma breakpoint region1; CEA, carcinoembryonic antigen; mo, month.  下载: 导出CSV
下载: 导出CSV
S2. Gene alteration in myoepithelioma of different sites
Tumor site Gene rearrangement Gene deficiency EWSR1 FUS OGT SRF PLAG1 RREB1 SMARCB1/ INI1 Soft tissue Yes[5] Yes[5] Yes[4, 6] Yes[7] Yes[8] Yes[9] Yes[10, 11] Bone Yes[5] Yes[12] NR NR NR NR NR Salivary gland No[5] NR NR NR Yes[13] NR NR Visceral locations (lung) Yes[5] Yes[5, 14] NR NR NR NR NR Skin Yes[15] NR NR NR Yes[16] NR NR MELTVR No[17] No[17] NR NR NR NR Yes[17] Note. Abbreviations: NR, not reported; MELTVR, myoepitheliom like tumors of the vulvar region; EWSR1, Ewing sarcoma breakpoint region1; FUS, fused in sarcoma gene; PLAG1, pleomorphic adenoma gene 1; OGT, O-linked N-acetylglucosamine; SRF, serum response factor; SMARCB, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1.
下载: 导出CSV
S3. Review of soft tissue myoepithelioma in children below 18 years olda
Authors No. of
casesAge/
sexSite Gene
rearrangementBehavior Invasive
growthRecurrence Metastasis Treatment/
outcomeKilpatrick et al, 1997[18] 1 9/M axilla NA Malignant* Yes Yes No Resection/
REC, 30 moWaldrop et al, 2001[19] 1 11/M thigh NA Malignant Yes No Orbir, 10 mo Resection/
died 4 mo after
metastasisVan den Berg
et al, 2004[20]1 17/M left lower leg NA Malignant NA − LN, L, bone, 2 mo amputationb/
died 7 mo after
amputationHarada et al,
2005[21]1 17/M forearm NA Malignant Yes No L, 6 mo Resection/
died 18 mo
after metastasisHallor et al,
2008[22]1 11/F back NA Benign Yes Yes, 6 y L, 7 y Resection/
AwD,9 yHerlihy et al,
2009[23]1 3mo/M orbit NA Benign No No No Resection/
NED, 36 moGuedes et al,
2009[24]1 6/F forearm NA Malignant No No L, 1 mo Resection+
chemo/, NAAntonescu et al, 2010[5] 7 9/M arm EWSR1 Benign NA NA NA NA 7/F hip EWSR1 Malignant NA NA NA NA 11/F forarm EWSR1 Malignant NA NA NA NA 13/F peri-ocular EWSR1 Malignant NA NA NA NA 7/F peri-orbital EWSR1 Malignant NA NA NA NA 1/M head&neck EWSR1 Malignant NA NA NA NA 2/F mediastinum EWSR1 Malignant NA NA NA NA Rekhi et al,
2012[25]1 18/M inguinal No Benign Yes NA NA NA Antonescu et al, 2013[17] 3 3mo/M knee No Malignant NA NA NA NA 3/M mediastinum No Malignant NA NA NA NA 8/F thigh No Malignant NA NA NA NA Park et al,
2013[26]1 18/M forearm NA Benign No No No Resection/
NED, 4 yGupta et al,
2016[27]1 2/F CNS NA Malignant Yes Yes Cervical
spinechemo+Resection+XRT/AwD, 30 mo Baldovini et al, 2018[28] 1 newborn neck NA Malignant No No No Resection+chemo+XRT/NED, 12 mo Bhanvadia et al, 2017[29] 1 14/M gluteal NA Benign No No No Resection/
NADe Cates et al, 2020[30] 1 15/M skull base No Benign No No No Resection/
NED, 24 moSegawa et al,
2020[8]2 16/M groin EWSR1 Benign No No No Resection/
NED, 24 mo5/F back EWSR1 Benign No No No Resection/
NED, 3 moTrevinoa et al, 2020[10] 1 12/M foot
plantarNA Malignant No NA L, pericardium, RP Resection/
NAThe present
case1 9/M mediastinum EWSR1 Benign Yes Yes, 6 mo No Resection/
AwD, 7 moNote. *Mitoses numbered eight per ten. aCases with ductal differentiation/structures were excluded. bTNF-α, melphalan, perfusion followed by amputation. Abbreviations: chemo, chemotherapy; NA, not available; NED, no evidence of disease; XRT, radiation therapy; AwD, alive with disease; LN, lymph node; L, lung; RP, retroperitoneum; REC, recurrence; CNS, central nervous system; mo, month; y, year.
下载: 导出CSV
-
[1] Jo VY, Doyle LA. Refinements in sarcoma classification in the current 2013 World Health Organization classification of tumours of soft tissue and bone. Surg Oncol Clin North Am, 2016; 25, 621−43. doi: 10.1016/j.soc.2016.05.001 [2] Brandal P, Panagopoulos I, Bjerkehagen B, et al. Detection of a t(1;22)(q23;q12) translocation leading to an EWSR1-PBX1 fusion gene in a myoepithelioma. Genes Chromosom Cancer, 2008; 47, 558−64. doi: 10.1002/gcc.20559 [3] Trevino M, Moorthy C, Kafchinski L, et al. Foot plantar soft tissue malignant myoepithelioma tumor: case report and review of the literature. Clin Imaging, 2020; 61, 90−4. doi: 10.1016/j.clinimag.2019.11.014 [4] Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol, 2003; 27, 1183−96. doi: 10.1097/00000478-200309000-00001 [5] Segawa K, Sugita S, Aoyama T, et al. Myoepithelioma of soft tissue and bone, and myoepithelioma-like tumors of the vulvar region: clinicopathological study of 15 cases by PLAG1 immunohistochemistry. Pathol Int, 2020; 70, 965−74. doi: 10.1111/pin.13017 [6] Ferretti C, Coleman H, Altini M, et al. Intraosseous myoepithelial neoplasms of the maxilla: diagnostic and therapeutic considerations in 5 South African patients. J Oral Maxillofac Surg, 2003; 61, 379−86. doi: 10.1053/joms.2003.50075 [7] Domingo-Musibay E, Oliveira AM, Okuno SH, et al. Myoepithelioma of soft tissues: a single institution retrospective case series. Am J Clin Oncol, 2018; 41, 357−61. doi: 10.1097/COC.0000000000000292 [8] Jo VY, Fletcher CDM. Myoepithelial neoplasms of soft tissue: an updated review of the clinicopathologic, immunophenotypic, and genetic features. Head Neck Pathol, 2015; 9, 32−8. doi: 10.1007/s12105-015-0618-0 [9] Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol, 2007; 31, 1813−24. doi: 10.1097/PAS.0b013e31805f6775 [10] Bisogno G, Tagarelli A, Schiavetti A, et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. Pediatr Blood Cancer, 2014; 61, 643−6. doi: 10.1002/pbc.24818 -
 21493+Supplementary Materials.pdf
21493+Supplementary Materials.pdf

-

 点击查看大图
点击查看大图
计量
- 文章访问数: 1136
- HTML全文浏览量: 535
- PDF下载量: 91
- 被引次数: 0

 Quick Links
Quick Links