

 下载:
下载:




-
Pneumoconiosis refers to a group of occupational lung diseases caused by prolonged inhalation and retention of harmful dust, primarily characterized by extensive lung fibrosis[1]. The classification of pneumoconiosis is based on the type of dust exposure and pathological characteristics, with the major subtypes including silicosis, asbestosis, coal workers’ pneumoconiosis (CWP), and other form[2]. The Global Burden of Disease study analyzed the incidence trends of various pneumoconioses across 195 countries and territories from 1990 to 2017, reporting a 66.0% increase in cases from 36,186 to 60,055, with notable regional variations[3]. The development of therapeutic drugs for pneumoconiosis remains challenging due to the inability to completely eliminate exposure sources, the irreversible nature of pulmonary fibrosis, and the unclear pathogenesis of the disease[4]. Therefore, a comprehensive investigation of pneumoconiosis pathogenesis and the development of innovative preventive and therapeutic strategies are essential.
The advancement of high-throughput sequencing, mass spectrometry analysis, and other technologies has facilitated the application of multi-omics approaches to obtain comprehensive disease insights. These approaches are instrumental in elucidating disease mechanisms, enabling drug repurposing, and advancing personalized medicine. With the emergence of the lung-gut axis concept, an increasing number of studies have focused on exploring the intricate interactions between gut microbiota, its metabolites, and pulmonary fibrosis[5,6]. At the transcriptomic and metabolomic levels, the reprogramming of the arachidonic acid metabolic pathway and its key metabolites, prostaglandin D₂ and thromboxane A₂, has been shown to significantly contribute to silicosis development, with rimatroban effectively mitigating the associated lung inflammation and fibrosis[7]. Furthermore, at the proteomic level, the phosphorylation of epidermal growth factor receptor and spleen tyrosine kinase has been identified as a potential therapeutic target in silicosis, with the targeted agents fostamatinib and gefitinib demonstrating efficacy in reducing inflammation and fibrosis, thereby improving lung function[8]. Additionally, studies suggest that DNA methylation plays a role in dust-induced pulmonary fibrosis, providing a theoretical basis for the potential application of DNA methyltransferase inhibitors in the treatment of CWP[9]. Therefore, this study investigates the associations between intestinal flora, proteome, genome, and gene methylation with pneumoconiosis risk, integrating multi-omics approaches to explore targeted diagnostic and therapeutic strategies for the disease.
Employing a Mendelian randomization approach and using genetic variation as an instrumental variable, this study aims to establish a causal link between multi-omics factors and pneumoconiosis risk[10]. Unlike traditional randomized controlled trials, Mendelian randomization (MR) analyses effectively minimize the impact of confounding factors and reverse causation, as genetic variants are randomly allocated at conception and remain unaffected by environmental influences or disease onset[11]. In this research, we integrated extensive genome-wide association study (GWAS) data with molecular quantitative trait loci (QTL) datasets using summary-data-based MR (SMR) to explore the causal connections between gut microbiota, proteins, gene expression, methylation levels, and the three subtypes of pneumoconiosis. To validate and complement the findings from the MR analysis, we analyzed the abundance and metabolic pathways of intestinal flora in patients with pneumoconiosis using 16S rRNA gene sequencing. Ultimately, by integrating multi-omics evidence of pneumoconiosis with existing drug molecule libraries, we provide new insights into identifying potential therapeutic agents for targeted treatments of the disease.
-
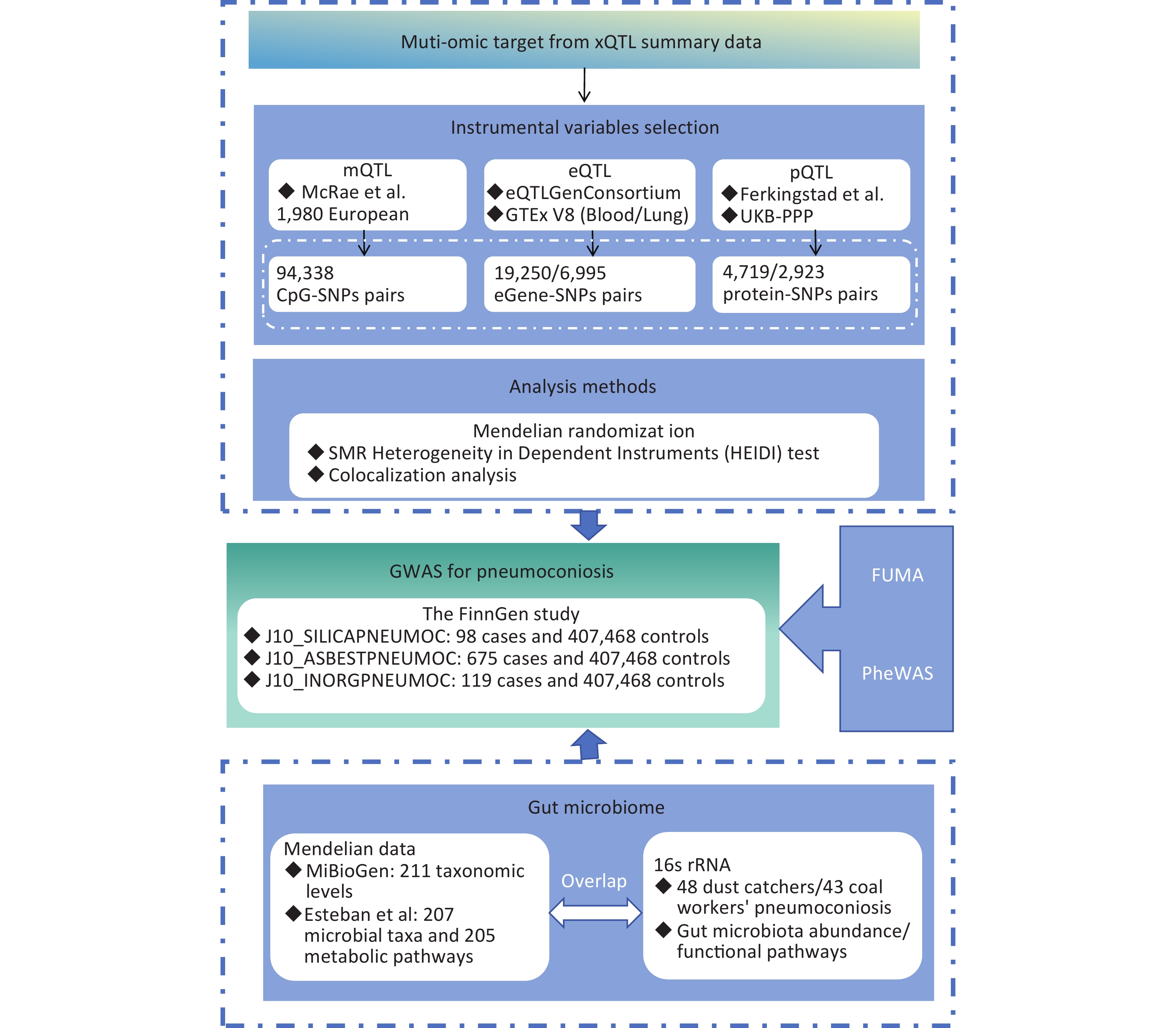
This study employs MR to synthesize evidence from gut microbiota and multi-omics data, aiming to expand personalized management options for pneumoconiosis patients. Figure 1 provides an overview of the study design. Our MR analysis primarily utilized publicly available datasets from the MiBioGen consortium, deCODE, UK Biobank Pharma Proteomics Project (UKB-PPP), the eQTLGen consortium, the Genotype-Tissue Expression (GTEx) portal, the FinnGen study, and research conducted by Esteban et al. and McRae et al. This MR study adheres to three fundamental assumptions: the relevance assumption, which requires that instrumental variables (IVs) be strongly associated with exposure factors; the independence assumption, which ensures that IVs are not influenced by known confounders; and the exclusion restriction assumption, which asserts that IVs affect the outcome solely through the exposure factor and not via alternative pathways[12].
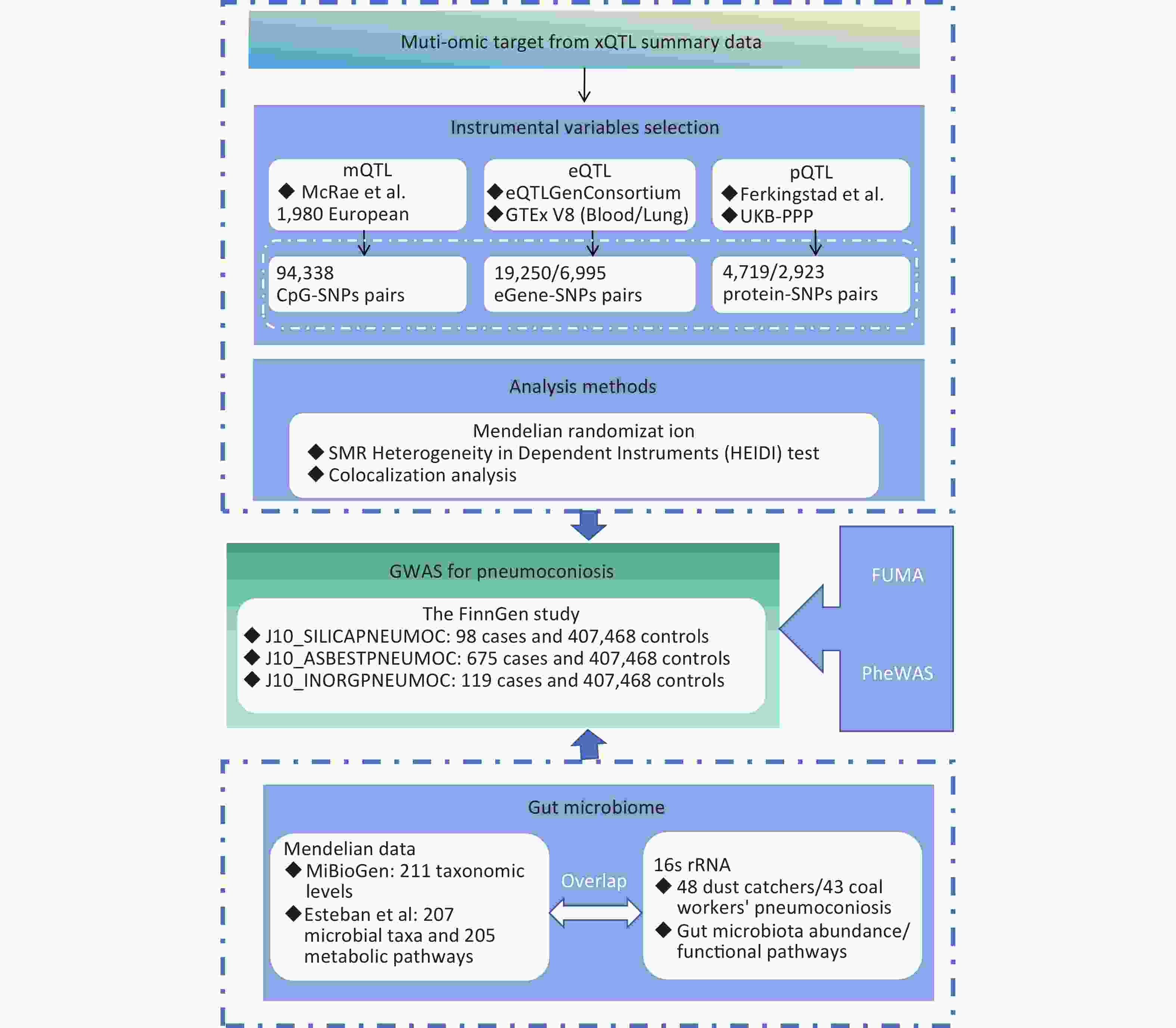
Figure 1. Study design for MR-based analysis of multi-omics and pneumoconiosis risk.
-
In this study, we used GWAS data for two gut microbiota datasets, obtained from the MiBioGen Consortium and the study by Esteban et al. The MiBioGen study (https://mibiogen.gcc.rug.nl/) included 24 cohorts comprising 18,340 participants, 85% of whom were of European ancestry Supplementary Table S1. This GWAS dataset explored 211 taxonomic levels, including 131 genera, 35 families, 20 orders, 16 classes, and 9 phyla. Additional data were obtained from a large-scale GWAS of 7,738 Dutch participants, which examined 207 microbial taxa and 205 metabolic pathways, providing insights into both the taxonomic composition and functional aspects of microbial communities[13].
-
The protein quantitative trait loci (pQTL) data utilized in this study were obtained from the deCODE Genetics and UKB-PPP datasets (https://registry.opendata.aws/ukbppp/). The team at deCODE Genetics conducted GWAS to assess plasma protein levels, analyzing data from 35,559 Icelandic participants and measuring 4,907 proteins[14]. UKB-PPP employed the Olink Explore™ platform and the Illumina NovaSeq™ 6000 sequencing platform to perform pQTL mapping, integrating proteomics and genomic sequencing data from 54,219 participants[15]. This large-scale analysis identified 14,287 significant genetic associations across 2,923 protein markers.
-
The eQTLGen Consortium and the GTEx project provided expression quantitative trait loci (eQTL) data to investigate how genetic variants influence gene expression and contribute to disease susceptibility. The GTEx project, which studies tissue-specific gene expression and regulatory mechanisms, has analyzed samples from 54 non-diseased tissue sites across nearly 1,000 individuals (https://www.gtexportal.org/). In this study, we specifically used eQTL data from peripheral blood and lung tissue to explore gene regulation in pneumoconiosis. Additionally, as a supplementary dataset, the eQTLGen consortium contributed cis-derived eQTL data from 31,684 individuals[16]. Given the critical role of gene methylation in transcriptional regulation, we incorporated data from methylation quantitative trait loci (mQTL) studies. We used 52,916 cis-mQTLs validated in the Brisbane Systems Genetics Study, which included 614 individuals from 177 families, along with the combined Lothian birth cohorts of 1921 and 1936, comprising 1,366 individuals[17]. These datasets provided essential insights into epigenetic modifications that may influence pneumoconiosis risk.
-
This study examined three types of pneumoconiosis—asbestosis, silicosis, and inorganic pneumoconiosis—using GWAS data from the FinnGen Biobank, which compiles and evaluates genomic and health data from hundreds of thousands of Finnish participants[18]. We used the R10 dataset, released on December 18, 2023, which includes data on silicosis (98 cases and 407,468 controls), asbestos-related pneumoconiosis (675 cases and 407,468 controls), and inorganic dust pneumoconiosis (119 cases and 407,468 controls). Diagnoses were based on the International Classification of Diseases 9 and 10 criteria. The cohort includes detailed classifications as follows: J61 and 501 correspond to pneumoconiosis caused by asbestos and other mineral fibers; J62 and 502 refer to silicosis, which results from inhalation of silica-containing dust; and J63 and 503 indicate pneumoconiosis caused by exposure to other inorganic dusts (inorganic pneumoconiosis).
-
To assess the causal relationship between variations in intestinal microbiota and QTL exposure, we employed basic MR and SMR methods.
-
The causal link between intestinal microbiota and pneumoconiosis was primarily investigated using the “TwoSampleMR” package. To select instrumental variables, we applied a P-value threshold of < 1 × 10−5 to ensure a strong association between single nucleotide polymorphisms (SNPs) and intestinal microbiota exposure. The 1000 Genomes European reference panel was used to calculate linkage disequilibrium (LD) between SNPs, maintaining an LD threshold of r² < 0.01. Additionally, we excluded SNPs with an allele effect frequency below 0.01 or those with a palindromic structure to preserve the integrity of instrumental variables. To mitigate the influence of weak instrumental variables, we required the F-statistic to exceed 10. The inverse variance weighting (IVW) method served as the primary approach for MR analysis, while the Wald ratio method was used for estimating individual SNP effects. MR-Egger and weighted median methods were employed as complementary approaches. To assess the robustness of the findings, Cochrane’s Q statistic was used to evaluate heterogeneity among SNPs, while pleiotropy was assessed using a combination of the MR-Egger intercept test, the global outlier test (MR-PRESSO), and leave-one-out analysis. The Steiger filter method was ultimately applied to confirm the directionality of the association between intestinal microbiota and pneumoconiosis, eliminating the impact of reverse causation on the outcomes.
-
To investigate the association between gene methylation, gene expression, protein abundance, and the risk of pneumoconiosis and its subtypes, we employed SMR, which provides enhanced statistical power. We adhered to stringent criteria when selecting instrumental variables and prioritized cis-QTLs due to their strong association with gene regulation and direct impact on gene expression. This biological directness minimizes confounding factors, thereby improving the precision and reliability of Mendelian randomization analysis. Cis-QTLs were identified by defining a window of ± 1,000 kb centered on the target gene and applying a P-value threshold of 1 × 10−5 for selection. To prevent sample mismatches and preserve the integrity of instrumental variables, we excluded variants where the allele frequency difference exceeded 0.2, thereby reducing potential confounding and bias. Pleiotropy was assessed using the heterogeneity in dependent instruments (HEIDI) test with a threshold of P < 0.01, and the results were integrated with SMR findings to improve the accuracy of gene expression-phenotype associations. We conducted colocalization analysis using the R package to identify causal variants shared by pneumoconiosis across mQTL, eQTL, and pQTL levels. Colocalization settings employed window thresholds of ± 1,000 kb for pQTL-GWAS and eQTL-GWAS, and ± 500 kb for mQTL-GWAS[19]. Evidence supporting colocalization was determined based on posterior probabilities of H4 (PPH4) exceeding 0.70.
-
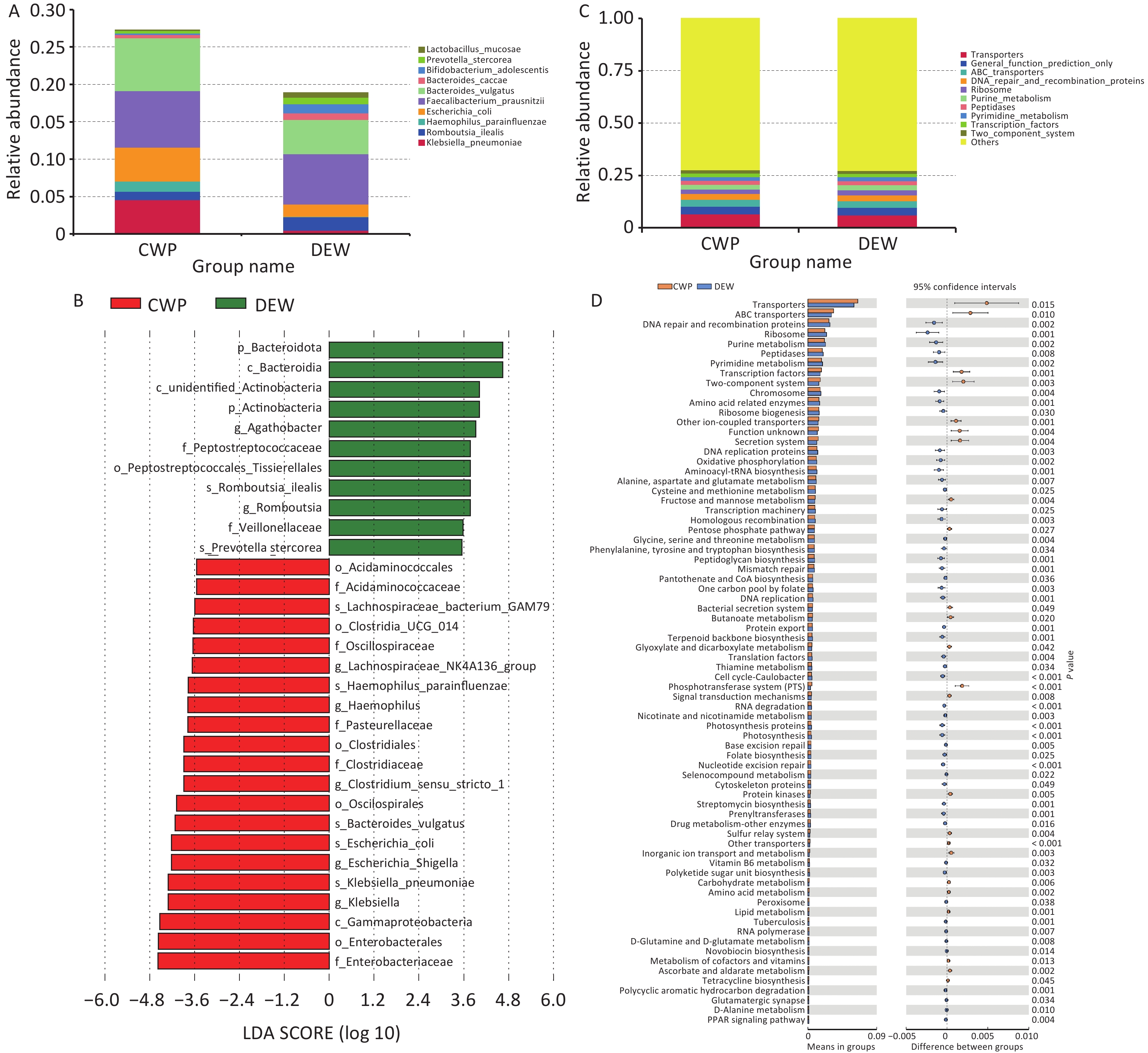
To further investigate the relationship between gut microbiota and pneumoconiosis, we recruited 43 male stage I CWP patients (CWP group) and 48 male dust-exposed workers (DEW group) for a study conducted by the NHC Key Laboratory of Pneumoconiosis at the First Hospital of Shanxi Medical University between October 2021 and August 2022. Inclusion criteria were as follows: (1) adult male participants, (2) a diagnosis of coal workers’ pneumoconiosis according to the GBZ70-2015 Diagnostic Criteria and the International Labour Organization Guidelines, and (3) for dust-exposed workers, a history of at least five years of coal dust exposure, absence of CWP symptoms, a normal lung condition confirmed via chest X-ray, and no indication of CWP based on diagnostic criteria. Exclusion criteria included (1) individuals with incomplete occupational history, (2) those with other respiratory diseases, and (3) individuals who had taken antibiotics within the past three months. Paired fecal samples were collected from participants for 16S rRNA gene sequencing to analyze and characterize gut microbial communities. Fresh fecal samples were obtained using sealed containers designed for fecal collection, with aseptic principles strictly followed throughout the collection process. Microbial species showing differential abundance between groups were analyzed using linear discriminant analysis effect size (LEfSe), focusing on operational taxonomic unit (OTU) clustering and species annotation to identify the top 10 taxa by relative abundance at each taxonomic level. Additionally, 16S rRNA sequencing data were processed using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) to infer functional compositional profiles, illustrating functional shifts in the gut microbiota of CWP patients (LDA > 3.5, P < 0.05). Details regarding ethical approval and clinical trial registration are provided in the supplementary online material.
-
We selected core SNPs associated with intestinal microbiota that corresponded to overlapping microbial taxa and functional pathways identified in both the MR analysis and our independent evaluation of gut microbiota in CWP patients. Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA) was employed, primarily using its SNP2GENE module (https://fuma.ctglab.nl/). This module refines potential candidate gene regions by identifying SNPs closely linked to core gut microbiota, facilitating fine mapping to pinpoint specific genes or gene variants that may influence the phenotype[20]. This approach enhances our understanding of how genetic variations affect biological traits and may reveal novel biological mechanisms or therapeutic targets for pneumoconiosis.
-
To elucidate the relationship between gut microbiota and QTL in pneumoconiosis risk, we integrated multi-omics data. At the gut microbiota level, taxa that remained significant after MR analysis and also demonstrated statistical significance in our independent assessment were classified as significantly associated microorganisms, while others were considered potentially associated. At the QTL level, genes that overlapped in at least two pQTLs and three eQTLs were categorized as significantly causally associated, whereas those with overlap in one pQTL and three eQTLs were designated as potentially causally associated. Additionally, genes and cis-eQTLs identified in FUMA using SNPs closely linked to core gut microbiota that also exhibited overlap with significant pQTLs were classified as additional genes with strong causal associations. A P-value < 0.05 was considered indicative of statistical significance.
-
Phenome-wide association studies (PheWAS) were employed to investigate the relationships between specific genetic variants and a broad range of phenotypes, providing insights into complex genetic and phenotypic interconnections. This study utilized the gene-based association scanning feature of ExPheWas (https://exphewas.statgen.org/) to analyze key genes. The analysis aimed to identify causal links between these genes and 26,616 phenotypes within the UK Biobank dataset, including 362 self-reported diseases, 21 manually defined cardiovascular endpoints, 1,280 phase sections, and 83 continuous variables[21]. All tests were adjusted for age, sex, and the first ten ancestry principal components, with association tests further stratified by sex.
-
To explore the potential of targeting key genes for pharmacological intervention, we used web-based pharmacological tools to assess the efficacy of known drugs and small-molecule compounds in treating pneumoconiosis. DrugBank (https://www.drugbank.com/) was employed to identify potential drug candidates that intersect with protein and gene expression data, selecting approved drugs for subsequent molecular docking analysis. Molecular docking, a critical computational technique in drug discovery and optimization, facilitates early-stage predictions of drug-like properties and informs further drug design and development. Protein structure data for drug candidates were obtained from the PubChem Compound Database (https://pubchem.ncbi.nlm.nih.gov/) and the Protein Data Bank (http://www.rcsb.org/). The binding affinities and interaction patterns between drug and small-molecule candidates and their respective targets were evaluated using AutoDock Vina 1.2.2, a computational protein-ligand docking software[22].
-
In the MiBioGen study, the number of SNPs used as IVs for each microbial taxon ranged from 3 to 21, totaling 2,353 SNPs included in the follow-up study. We identified 2, 9, and 10 gut microbial taxa associated with the risk of asbestosis, inorganic pneumoconiosis, and silicosis, respectively. Among the protective factors against asbestosis, we identified Dorea, Eubacterium rectale group, Clostridia class, Butyricicoccus genus, and Marvinbryantia genus. Conversely, Eubacterium ruminantium group, Lachnospira genus, Butyrivibrio genus, and Alistipes genus were associated with an increased risk of asbestosis. Regarding inorganic pneumoconiosis, elevated levels of Marvinbryantia and Olsenella genera were linked to a decreased disease risk, while Catenibacterium, Holdemania, and Dialister genera were associated with an increased risk. Similarly, increased levels of Methanobrevibacter genus, Methanobacteria class, Methanobacteriales order, and Methanobacteriaceae family, alongside Marvinbryantia genus, were found to reduce the risk of inorganic pneumoconiosis. Conversely, increased levels of Catenibacterium, Holdemania, and Dialister were correlated with a higher disease risk. In the context of silicosis, genus.unknowngenus.id.1868, Dialister genus, and Romboutsia genus, all classified under Erysipelotrichales order, Erysipelotrichaceae family, and Erysipelotrichia class, exhibited a positive correlation with disease risk. In contrast, Adlercreutzia genus, Ruminococcaceae UCG002 genus, Mollicutes class, and Tenericutes phylum demonstrated a negative correlation with silicosis risk (Figure 2 and Supplementary Tables S2–S4). Similarly, in an independent analysis, we employed a total of 3,974 SNPs as instrumental variables, with the number of strongly associated SNPs per microbial colony ranging from 3 to 19. After excluding weak instrumental variables, the mean F-statistic value was 21.56. We identified 10 functional pathways and 10 gut microbial abundances causally linked to asbestosis, 10 pathways and 12 gut microbial abundances associated with inorganic pneumoconiosis, and 8 pathways and 4 gut microbial abundances causally linked to silicosis (Supplementary Tables S5–S7).

Figure 2. Associations between genetically predicted Gut microbes and pneumoconiosis in Mendelian randomization analysis (A: Esteban et al., B: MiBioGen Consortium).
-
Following the application of rigorous screening criteria, we identified 94,338 CpG-SNP pairs from McRae et al., 19,250/6,995 eGene-SNP pairs from the eQTLGen Consortium and GTEx V8 (Blood/Lung), and 4,719/2,923 protein-SNP pairs from Ferkingstad et al. (UKB-PPP to serve as instrumental variables for SMR analysis. We considered a result statistically significant if it met the thresholds of P < 0.05 and P-HEIDI < 0.01.
-
In our investigation of asbestosis, we identified causal associations with 750 genes in eQTLGen, 313 genes in lung tissue, and 269 eQTLs in peripheral blood, with 70 eQTLs overlapping across all three datasets (Supplementary Tables S8–S10). Subsequently, we identified 81 pQTLs from the deCODE dataset and 102 pQTLs from UKB-PPP, both associated with the risk of asbestosis (Supplementary Tables S11–S12). Ultimately, five overlapping genes—GGPS1, STX8, NT5C3B, ACADM, and GSTM4—were identified. Among these, STX8 was found to overlap in two protein datasets, GSTM4 was observed at methylation site cg05793930, ACADM at cg05467918, and NT5C3B exhibited methylation at cg13092355 and cg26502583 (Supplementary Table S13).
-
Similarly, we identified 618, 242, and 249 eQTLs across the three eQTL datasets that were significantly associated with inorganic pneumoconiosis. An intersection analysis of these datasets revealed 70 key eQTLs (Supplementary Tables S14–S16). From the two pQTL data sources, we identified 62 and 90 pQTLs that were causally associated with the risk of inorganic pneumoconiosis (Supplementary Tables S17–S18). Ultimately, PPT1 and FBN2 were found to influence inorganic pneumoconiosis across eQTL, pQTL, and mQTL levels, specifically at methylation sites cg04560534, cg22716190, cg02271895, and cg17564775. Additionally, MIF was consistently confirmed in two pQTL and three eQTL datasets, with methylation site cg10819733 being particularly significant (Supplementary Table S19).
-
Applying the same methodology to establish the causal relationship between QTL and silicosis, we identified a total of 74 eQTLs from three intersecting databases, along with 74 pQTLs from the deCODE repository and 72 pQTLs from UKB-PPP, all strongly associated with the risk of developing silicosis (Supplementary Tables S20–S24). In our final analysis, we discovered that GM2A and TPK1 functioned as protective factors against silicosis, exerting beneficial effects at both the genetic and protein levels. Additionally, methylation at cg21370481 and cg23307858 further enhanced TPK1 expression, contributing to a reduced risk of disease (Supplementary Table S25). Despite incorporating multiple sources of eQTL and pQTL overlaps to improve the likelihood of colocalization, we observed that UBP8 exhibited the highest PPH4 value, which was only 0.46 (Supplementary Table S26–S27).
-
Using an OTU clustering approach, we identified the 10 most abundant fecal bacterial species, which clustered into two primary groups. These species included Klebsiella pneumoniae, Romboutsia ilealis, Haemophilus parainfluenzae, Faecalibacterium prausnitzii, Escherichia coli, Bacteroides vulgatus, Bacteroides caccae, Bifidobacterium adolescentis, Prevotella stercorea, and Lactobacillus mucosae (Supplementary Table S28). Furthermore, we conducted LEfSe analysis to examine gut microbiota differences between dust-exposed workers and CWP patients, identifying 31 distinct microbial differentiation levels (Figure 3). The detailed results are presented in Supplementary Table S29. To characterize functional alterations in the gut microbiota of CWP patients, we analyzed 16S rRNA sequencing data using PICRUSt, predicting the top 10 functional categories within the compositional profile. These categories included transporters, general function prediction only, ABC transporters, DNA repair and recombination proteins, ribosome, purine metabolism, peptidases, pyrimidine metabolism, transcription factors, and two-component systems (Supplementary Tables S29–S30). The Supplementary Tables S28–S31 provide comprehensive data on functional differences in microbiota composition.
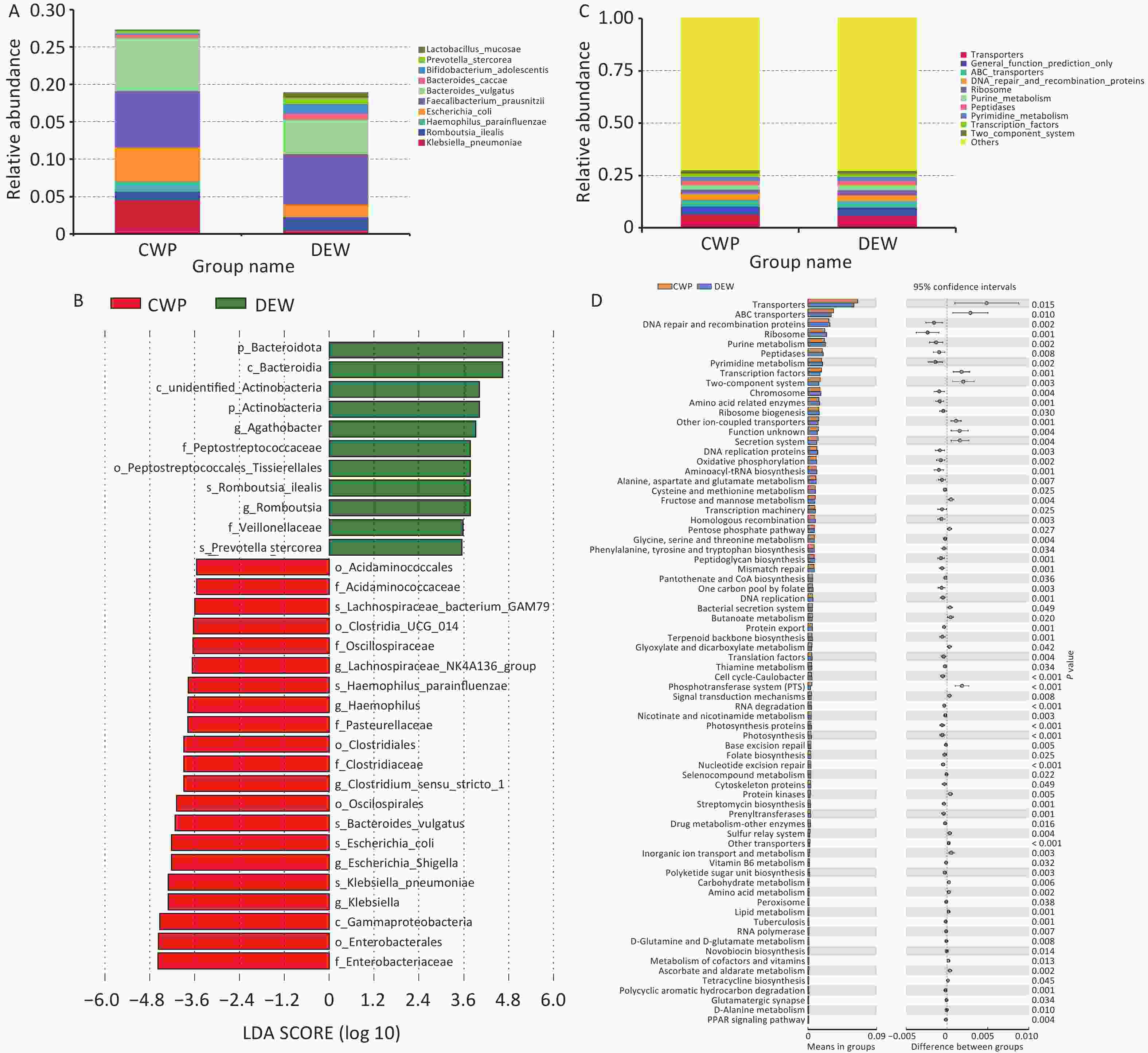
Figure 3. Characterization of gut microbiota in self-assessed pneumoconiosis cohort. (A) Top 10 most abundant gut microbial species; (B) Relative abundance differences in microbial species between groups; (C) Top 10 functional classes of gut microbiota; (D) Differences in gut microbial functional pathways.
-
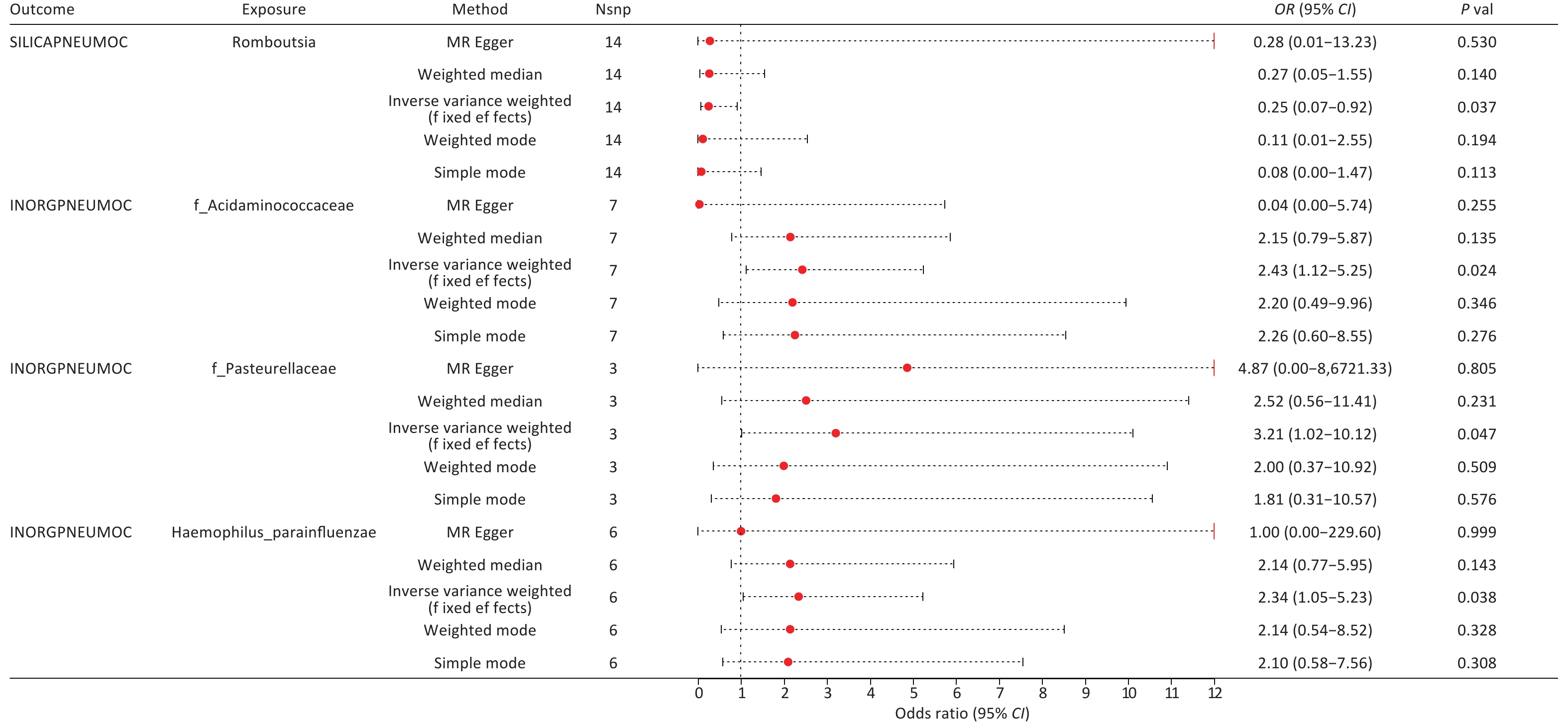
By integrating the significant findings from the MR analyses of two gut microbial taxa and three types of pneumoconiosis with the statistically significant microbial species identified in our independent assessment, we successfully identified four common microbial taxa. These included Haemophilus parainfluenzae, which ranked among the top 10 in relative abundance at the species level, along with Romboutsia genus (g_Romboutsia), Acidaminococcaceae family (f_Acidaminococcaceae), and Pasteurellaceae family (f_Pasteurellaceae), all of which were found within the differentially abundant microbiota (Figure 4 and Supplementary Table S32). Among these taxa, we ultimately selected only three core groups, as the results for f_Acidaminococcaceae showed inconsistent directionality when analyzed using both IVW and MR-Egger methods (Figure 5). The 23 SNPs associated with these four microbiota were linked to 50 essential genes and 27 cis-eQTLs (Supplementary Tables S33–S34). Notably, we found that the Vimentin (VIM) gene, identified from the deCODE dataset, was causally linked to asbestosis and simultaneously intersected with mapped genes and cis-eQTLs. Consequently, we identified VIM as a pivotal gene for asbestosis.
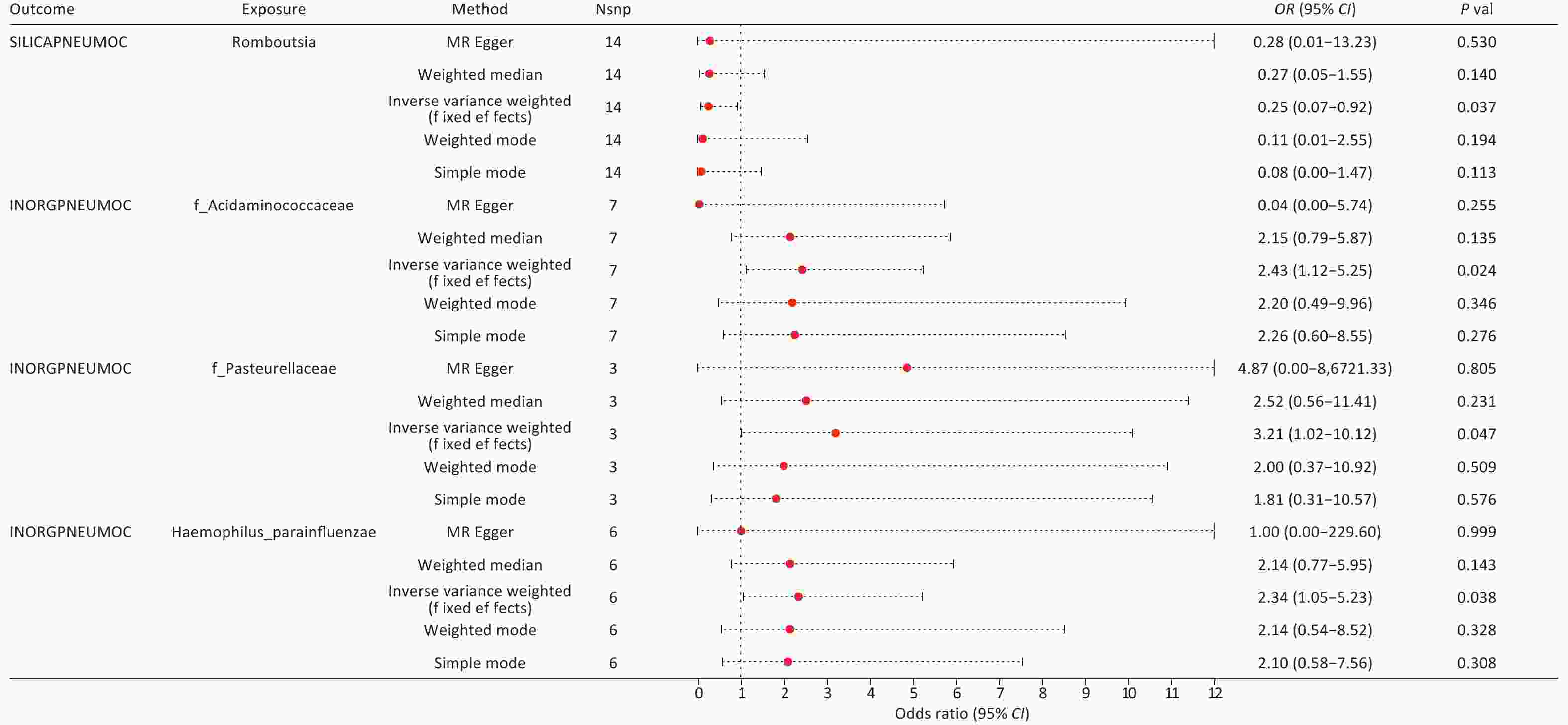
Figure 4. Forest plot of genetically predicted four overlapping gut microbes associated with pneumoconiosis in Mendelian randomization analysis.
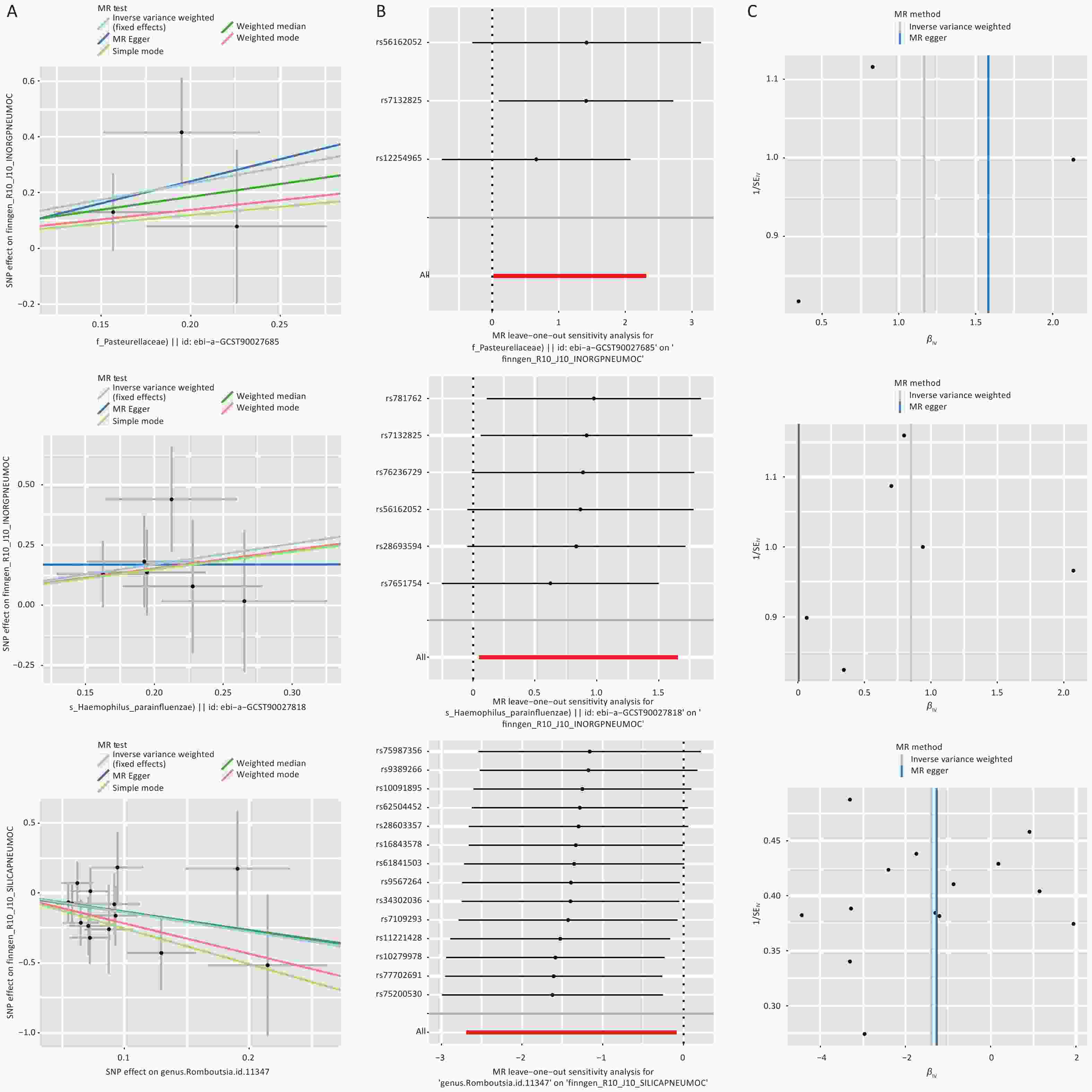
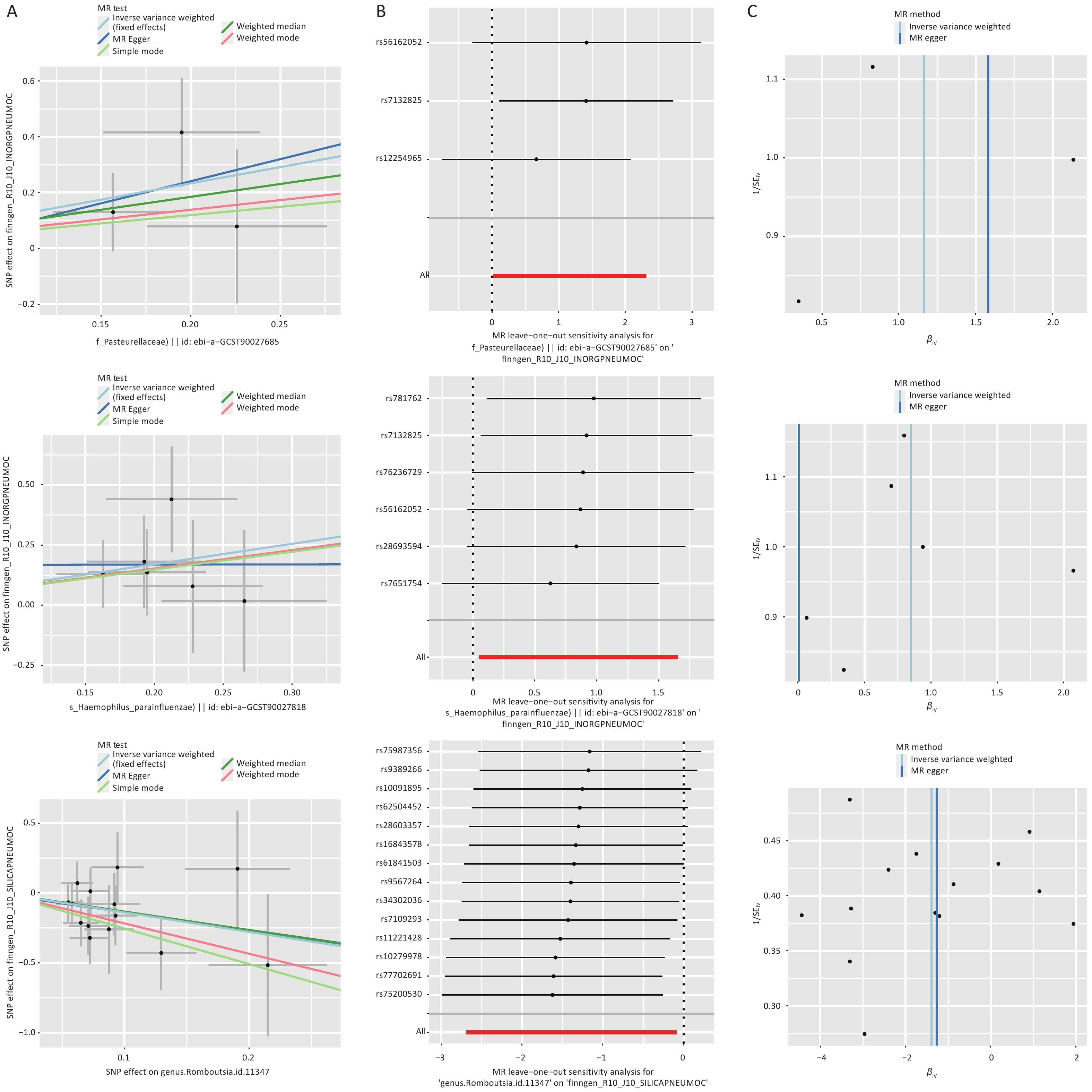
Figure 5. Statistical analyses of three core gut microbiota taxa. (A) Scatter plot; (B) Leave-one-out (LLO)plot; (C) funnel plot.
-
Upon integrating multi-omics data, we identified strong causal associations by aligning the positively correlated taxa from MR analyses with statistically significant microbial taxa from our independent assessment, which included s_Haemophilus parainfluenzae, g_Romboutsia, f_Acidaminococcaceae, and f_Pasteurellaceae. At the QTL level, we pinpointed genes causally linked to pneumoconiosis subtypes, including those significantly associated with asbestosis (STX8, VIM) and potentially associated with GGPS1, NT5C3B, GSTM4, and ACADM. For inorganic pneumoconiosis, MIF was significantly associated, while PPT1 and FBN2 were potentially associated. In silicosis, we identified GM2A and TPK1 as potential candidate genes. In total, we identified 10 genes with causal relationships to pneumoconiosis (Table 1 and Supplementary Table S35).
Table 1. In Mendelian randomization analyses, the genes of candidate genes predict methylation, expression, and proteins in relation to pneumoconiosis
Outcome Gene Tier mQTL eQTL (eQTLGen) eQTL (GTEx Lung) eQTL (GTEx blood) pQTL (deCODE) pQTL (UKBppp) Probe OR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueAsbestosis STX8 significant 1.383
(1.037–1.844)0.027 1.735
(1.037–2.902)0.036 2.054
(1.046–4.034)0.037 8.663
(1.161–64.631)0.035 4.23
(1.151–15.545)0.03 VIM significant 2.593
(1.508–4.459)0.001 0.191
(0.041–0.887)0.035 GGPS1 potential 1.64
(1.04–2.58)0.033 1.996
(1.033–3.86)0.04 2.036
(1.032–4.02)0.04 4.433
(1.085–18.115)0.038 NT5C3B potential cg13092355 0.87
(0.768–0.973)0.029 1.14
(1.013–1.283)0.029 1.149
(1.011–1.305)0.033 1.141
(1.013–1.285)0.029 1.228
(1.021–1.477)0.029 GSTM4 potential cg05793930 0.614
(0.415–0.814)0.015 0.815
(0.697–0.953)0.011 0.801
(0.678–0.945)0.009 0.855
(0.761–0.96)0.008 0.885
(0.702–1.116)0.301 0.779
(0.432–1.404)0.407 ACADM potential cg05467918 1.287
(1.011–1.563)0.04 0.694
(0.494–0.976)0.036 0.486
(0.241–0.981)0.044 0.576
(0.338–0.981)0.042 0.581
(0.336–1.005)0.052 0.402
(0.168–0.964)0.041 Inorganic pneumoconiosis MIF significant cg10819733 0.556
(0.334–0.778)0.024 4.559
(1.199–17.333)0.026 1.343
(1.036–1.739)0.026 1.412
(1.043–1.913)0.026 0.014
(0.00023–0.816)0.04 0.052
(0.004–0.612)0.019 PPT1 potential cg04560534 4.23
(1.067–7.393)0.04 0.688
(0.493–0.959)0.028 8.131
(1.038–63.727)0.046 0.482
(0.251–0.925)0.028 9.11
(1.774–46.782)0.008 FBN2 potential cg02271895 3.467
(1.282–5.653)0.014 0.637
(0.429–0.945)0.025 0.681
(0.485–0.956)0.026 0.569
(0.346–0.934)0.026 0.351
(0.141–0.879)0.025 Silicosis GM2A potential 0.676
(0.462–0.989)0.044 0.232
(0.054–0.999)0.05 0.688
(0.478–0.99)0.044 0.271
(0.076–0.968)0.044 1.229
(0.803–1.881)0.341 TPK1 potential cg21370481 1.57
(1.131–2.01)0.007 0.49
(0.272–0.886)0.018 0.305
(0.108–0.864)0.025 0.223
(0.062–0.797)0.021 0.389
(0.177–0.852)0.018 Note. mQTL: Methylation Quantitative Trait Locus; eQTL: Expression Quantitative Trait Locus; pQTL: Protein Quantitative Trait Locus; OR: Odds Ratio; STX8: Syntaxin 8; VIM: Vimentin; GGPS1: Geranylgeranyl Diphosphate Synthase 1; NT5C3B: 5’-Nucleotidase Cytosolic III B; GSTM4: Glutathione S-Transferase Mu 4; ACADM: Acyl-CoA Dehydrogenase Medium Chain; MIF: Macrophage Migration Inhibitory Factor; PPT1: Palmitoyl-Protein Thioesterase 1; FBN2: Fibrillin 2; GM2A: GM2 Ganglioside Activator; TPK1: Thiamine Pyrophosphokinase 1. -
Using the ExPheWas online tool, we linked 10 genes to 362 self-reported diseases, 21 manually curated cardiovascular endpoints, 1,280 coherence degrees, and 83 continuous variables. After applying Bonferroni correction, we observed that the continuous variables were predominantly related to lung function (forced expiratory volume in one second), lipid levels (cholesterol, body fat percentage), blood parameters (red blood cell [erythrocyte] distribution width), and sex hormone-binding globulin (SHBG). Among self-reported conditions, we identified high cholesterol, hypertension, and cardiovascular diseases as the most frequently reported. Additionally, phecodes showed strong associations with hyperlipidemia, hypercholesterolemia, and essential hypertension (Supplementary Table S36).
-
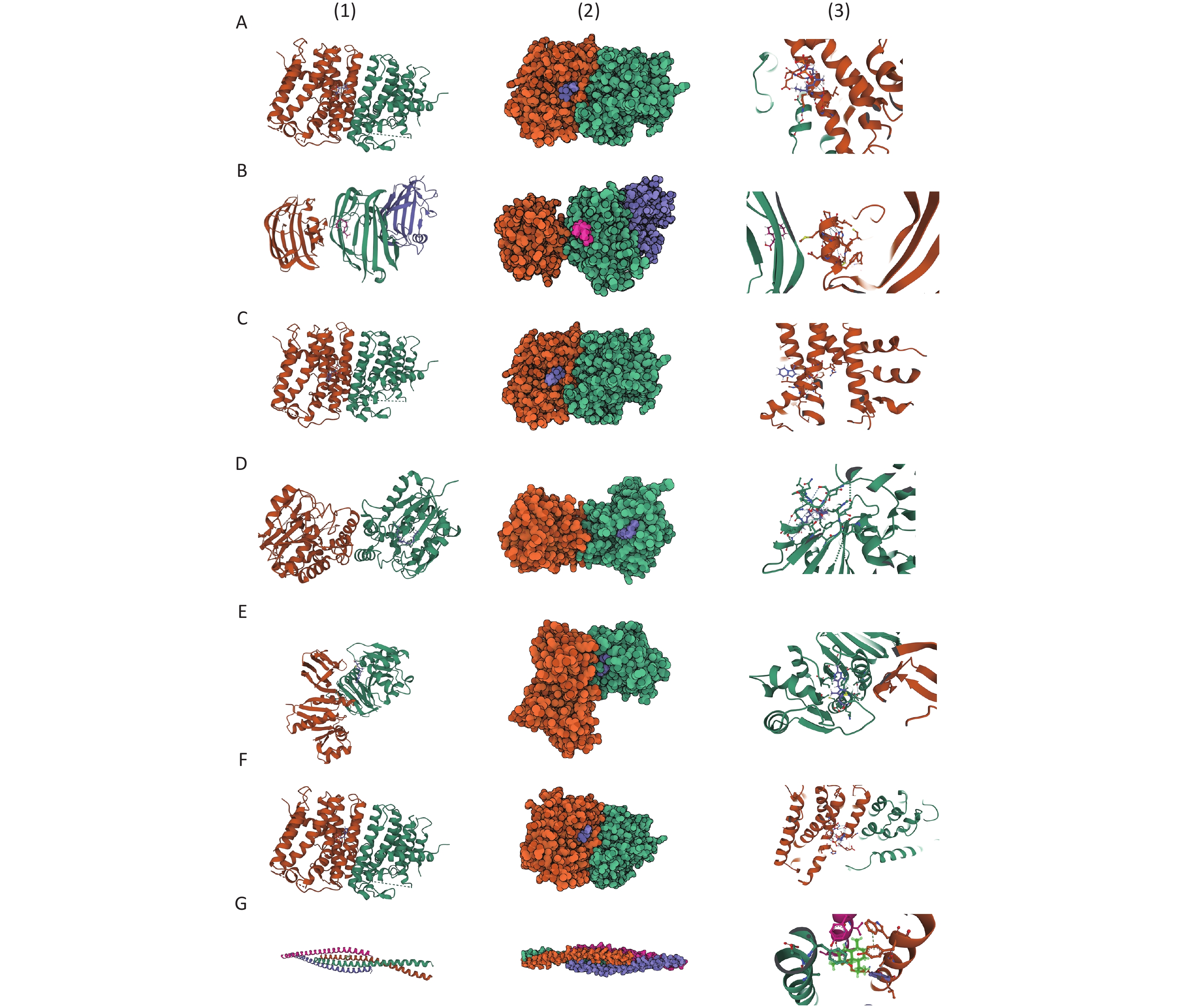
For asbestosis, Zoledronic acid, Minodronic acid, and Ibandronate were identified as inhibitors of GGPS1, while Glutathione and Flavin adenine dinucleotide were found to target GSTM4 and ACADM, respectively. Additionally, Artenimol was identified as a ligand for the VIM protein. For inorganic pneumoconiosis, Palmitic acid was found to target PPT1, while Calcium citrate, Calcium phosphate, and Calcium phosphate dihydrate functioned as ligands for FBN2. In the case of silicosis, Lauric acid was identified as a target for GM2A, and Thiamine acted as a substrate for TPK1. The binding configurations and molecular interactions of the 14 drug candidates with eight proteins are illustrated in Figure 6 and Supplementary Table S37, where the binding energies for these interactions are also listed.
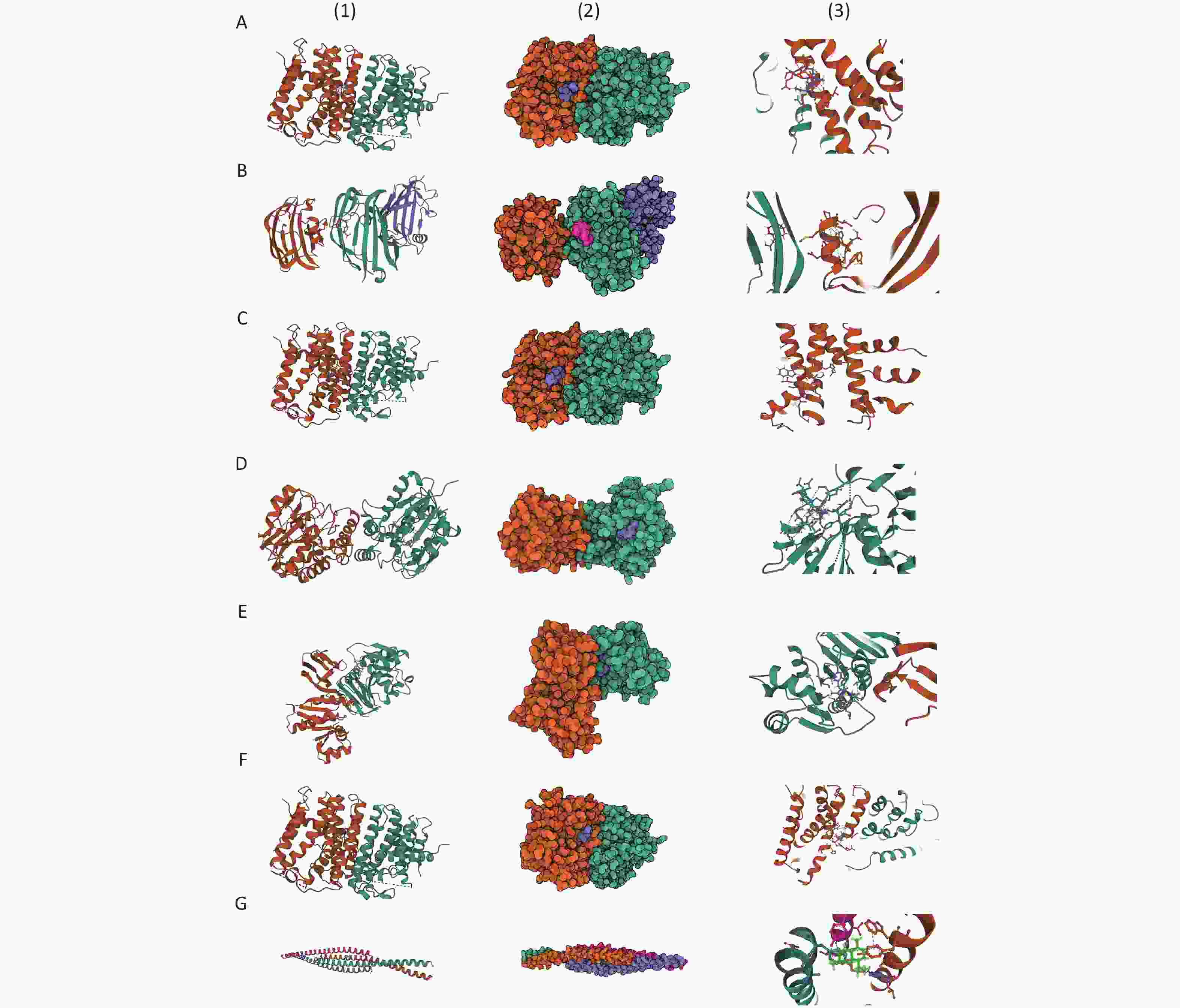
Figure 6. Prediction of potential drugs candidates and molecular docking analysis. (A) GGPS1-Zoledronic acid; (B) GGPS1-Minodronic acid; (C) GGPS1-Ibandronate; (D) PPT1-Palmitic Acid; (E) GM2A-Lauric acid; (F) TPK1-Thiamine; (G) VIM-Artenimol.
-
In this study, we explored, for the first time, the causal relationships between two gut microbiota taxa, six QTLs, and three types of pneumoconiosis using a combination of conventional MR and SMR methods. Additionally, we analyzed 16S rRNA gene sequencing data from fecal samples of pneumoconiosis patients and dust-exposed workers as an external validation method. This analysis revealed that Romboutsia acts as a protective factor against silicosis, while Pasteurellaceae and Haemophilus parainfluenzae were identified as risk factors for inorganic pneumoconiosis. At the QTL level, our findings suggest that elevated STX8 expression may increase the risk of asbestosis, whereas higher in vivo levels of MIF could reduce the risk of inorganic pneumoconiosis. Furthermore, GM2A and TPK1 were identified as protective factors against silicosis. Additionally, we examined four core gut microbiota taxa associated with key asbestosis-related genes and cis-eQTLs, including VIM, using FUMA. This study provides novel insights into early pneumoconiosis prevention and potential mitigation strategies by integrating gut microbiota and QTL data within a multi-omics framework. By elucidating the interplay between gut microbiota and genetic factors, our findings may help reduce disease progression and improve patient outcomes.
By integrating findings from MiBioGen, Esteban et al., and our own gut microbiota analysis, we identified Pasteurellaceae and Haemophilus parainfluenzae as potential risk factors for inorganic pneumoconiosis. Notably, our results revealed that Pasteurellaceae levels in the intestinal microbiota were significantly higher in CWP patients than in dust-exposed workers. Conversely, Romboutsia was identified as a potential protective factor against asbestosis, exhibiting a decreasing trend in the gut microbiota of pneumoconiosis patients. The Pasteurellaceae family encompasses a diverse range of Gram-negative bacteria, primarily inhabiting the respiratory, gastrointestinal, and reproductive tracts of animals. Within this family, the genus Haemophilus has been strongly associated with pulmonary fibrosis, cystic fibrosis, and chronic respiratory infections in humans[23,24]. Notably, unencapsulated Haemophilus influenzae has been implicated in acute exacerbations of pulmonary fibrosis by significantly inducing interleukin-17 (IL-17) secretion, potentially highlighting its role as a risk factor in the progression of pneumoconiosis[25,26]. Haemophilus parainfluenzae, a commensal of the human respiratory tract, exhibits ambiguous pathogenic potential compared to Haemophilus influenzae. Research suggests that compromised airway defenses, which delay bacterial clearance, may allow H. parainfluenzae to become pathogenic in the lower respiratory tract, potentially leading to severe infections such as pneumonia and bronchitis[27]. Cohort data indicate that in pneumoconiosis patients, H. parainfluenzae ranks fifth in abundance (1.19%) within the intestinal microbiota, significantly exceeding its levels in dust-exposed workers. This discrepancy may arise from the fact that chronic lung inflammation and fibrosis in pneumoconiosis patients create a favorable environment for bacterial colonization and proliferation. Additionally, Roseburia, a key gut microbiota member, constitutes 2%–31% of the total bacterial population in healthy individuals and is known to produce short-chain fatty acids—including acetic acid, propionic acid, and butyric acid—through the fermentation of dietary carbohydrates[28]. Among these, butyrate has demonstrated anti-fibrotic effects on fibroblasts, both directly and indirectly, by modulating macrophage differentiation and inhibiting histone deacetylase 3. Furthermore, butyrate may help mitigate pulmonary fibrosis by enhancing the body’s anti-inflammatory response[29].
Utilizing SNPs from three core gut microbiota species, we identified key genes using FUMA. Our analysis revealed that VIM exhibited overlapping associations across mapping genes, cis-eQTLs, and pQTLs within the deCODE dataset, as well as eQTL associations in the eQTLGen database. Vimentin, a type III intermediate filament protein, serves as a structural anchor supporting intracellular organelles in mesenchymal cells. Its expression is notably upregulated during epithelial-mesenchymal transition (EMT), a crucial process involved in lung fibrosis, wound healing, and cancer metastasis[30]. Research has demonstrated that asbestos exposure induces EMT in MeT-5A cells by downregulating epithelial markers—including E-cadherin, β-catenin, and Occludin—while upregulating mesenchymal markers such as fibronectin, α-smooth muscle actin (α-SMA), and vimentin. Moreover, EMT activation in alveolar epithelial cells accelerates the progression of pulmonary fibrosis[31]. Conversely, Vimentin also functions as a ligand for NOD-like receptor protein 3 (NLRP3) inflammasomes, facilitating inflammation and EMT in lung fibrosis through the NF-κB/NLRP3 signaling pathway. The observed decrease in epithelial markers alongside the increase in mesenchymal markers, including Vimentin, during EMT highlights Vimentin as a promising early biomarker for lung fibrosis-related conditions, such as pneumoconiosis.
Syntaxin 8 (STX8), a critical component of the endosomal SNARE complex, which includes synaptic binding protein 7, vesicle transport through interaction with t-SNAREs homolog 1B, and vesicle-associated membrane protein 8, has been identified as a risk factor for asbestosis across all QTL datasets[32]. Research has shown that overexpression of Syntaxin 8 disrupts the transport and function of the cystic fibrosis transmembrane conductance regulator, resulting in mucus stasis, chronic bacterial infections, and sustained inflammatory responses in the airways and lungs[33]. These pathological changes collectively exacerbate lung damage and impair ciliary function, thereby reducing the lungs’ ability to expel inhaled particles. This impaired clearance mechanism disrupts pulmonary homeostasis, posing a particular risk for pneumoconiosis patients and potentially contributing indirectly to the progression of pulmonary fibrosis[34].
Migration inhibitory factor (MIF) is a complex and highly debated cytokine that appears to have a dual role in pathological conditions. In this study, MIF was identified as a protective factor against inorganic pneumoconiosis at the pQTL level, yet paradoxically emerged as a risk factor at eQTL level. This conflicting evidence necessitated a detailed examination of MIF’s specific mechanisms. A study investigating bleomycin-induced pulmonary fibrosis in rats demonstrated that MIF knockdown alleviated lung damage and reduced extracellular matrix accumulation. This intervention notably decreased the levels of transforming growth factor-β1 (TGF-β1), tumor necrosis factor-α (TNF-α), IL-17, hydroxyproline, fibroblast growth factor 23, and secreted phosphoprotein 1 (Spp1). Additionally, MIF knockdown suppressed the expression of CD68, F4/80, and α-SMA proteins, effectively mitigating lung inflammation and fibrosis[35]. Moreover, MIF knockdown has been implicated in pulmonary fibrosis treatment, primarily through two mechanisms: (1) inhibiting fibrosis via the TGF-β1/Smads signaling pathway, and (2) reducing vascular remodeling by downregulating thrombospondin-2 (Thbs2) and serpin family B member 5 mRNA expression. Conversely, MIF also plays a crucial role in coordinating both innate and adaptive immune responses, contributing to the suppression of host inflammatory reactions to invading pathogens[36]. Increasing evidence suggests that MIF actively modulates macrophage responses during infections by promoting the production of pro-inflammatory cytokines and other inflammatory mediators, including TNF-α, interferon-gamma, interleukin-1β, interleukin-2, interleukin-6, interleukin-8, nitric oxide, cyclooxygenase-2, and several matrix metalloproteinases and their inhibitors[37-39]. Furthermore, the methylation of cg10819733 has been suggested to increase MIF expression, potentially influencing pneumoconiosis risk. This observation underscores the need for further research into MIF’s intricate and context-dependent role in pneumoconiosis pathogenesis.
This study represents the first MR analysis to integrate protein expression, gene expression, gene methylation, and gut microbiota composition to investigate their potential associations with pneumoconiosis risk. To identify the most pivotal genes, we utilized a comprehensive multi-omics approach, incorporating pQTL data from deCODE and UKB-PPP, expression eQTL data from eQTLGen, the GTEx database (blood and lung tissue), and mQTL data from McRae et al. For gene and cis-eQTL mapping, we integrated findings from MiBioGen, Esteban et al., and our intestinal microbiota analysis, subsequently superimposing pQTL data to refine gene selection. Additionally, by comparing asbestosis, inorganic pneumoconiosis, and silicosis at a multi-omics level, we aimed to provide insights into personalized disease management and improve targeted prevention and treatment strategies for pneumoconiosis.
Additionally, we assessed the limitations of our study. Due to data source constraints, the maximum number of pneumoconiosis-related cases included in our analysis was limited to 675 patients, requiring caution when generalizing these findings to broader populations. Furthermore, our external validation of pneumoconiosis cases was conducted in an Asian cohort, which may yield different outcomes compared to European populations. Moreover, we were unable to identify a fully aligned overlap between the MR analysis and cohort-based microbial functional pathways, a discrepancy that may stem from methodological inconsistencies. This study exclusively included CWP patients, considering their antibiotic use, but did not account for the potential effects of immunomodulatory drugs, hormonal therapies, antifibrotic agents, or nutritional supplements on intestinal microbiota composition, all of which could influence the results. In selecting IVs, we prioritized securing adequate IVs for further research, opting for a P-value threshold of < 1 × 10−5 instead of the more stringent threshold of 5 × 10−8, acknowledging that our findings may be more susceptible to false positives. Furthermore, while we selected intersecting positive results from one mQTL dataset, two pQTL datasets, and three eQTL datasets to identify the most significant genes, the peak posterior probability for hypothesis 4 (PPH4) value in co-localization analysis did not reach the robust statistical threshold (PPH4 = 0.7). In conclusion, while our study provides statistical insights into the potential pathogenesis of pneumoconiosis, further validation through animal and cellular studies is essential to translate these findings into therapeutic applications for the disease.
-
This study employed a MR approach to investigate potential causal relationships and explore novel biological mechanisms linking multi-omics data and gut microbiota to pneumoconiosis risk. Using SMR, we integrated data from three eQTL and two pQTL sources, identifying two genes (STX8 and MIF) significantly associated with pneumoconiosis risk and highlighting eight additional genes with potential associations. Furthermore, our findings indicated that methylation of cg10819733 influences MIF expression, potentially impacting pneumoconiosis susceptibility. By synthesizing publicly available microbiota data alongside Independent-assessment from a pneumoconiosis cohort, we identified three core gut microbiota taxa—Pasteurellaceae, Haemophilus parainfluenzae, and Romboutsia—associated with pneumoconiosis. Through fine-mapping of genes, cis-eQTL analysis, and previous multi-omics evidence, VIM was identified as an additional significantly associated gene. Subsequently, we established associations between key genes and multiple phenotypic traits, uncovering strong links with lung function, lipid levels, blood parameters, and SHBG. These factors may serve as complementary contributors to pneumoconiosis pathogenesis. Additionally, by applying predictive modeling and molecular docking approaches, we identified potential therapeutic agents targeting these genes and pathways. Overall, this study provides new insights into the development of personalized therapeutic strategies for pneumoconiosis and highlights potential drug targets for screening and repurposing, offering new directions for disease management and treatment optimization.
doi: 10.3967/bes2025.028
Deciphering the Role of VIM, STX8, and MIF in Pneumoconiosis Susceptibility: A Mendelian Randomization Analysis of the Lung-Gut Axis and Multi-Omics Insights from European and East Asian Populations
-
Abstract:
Objective Pneumoconiosis, a lung disease caused by irreversible fibrosis, represents a significant public health burden. This study investigates the causal relationships between gut microbiota, gene methylation, gene expression, protein levels, and pneumoconiosis using a multi-omics approach and Mendelian randomization (MR). Methods We analyzed gut microbiota data from MiBioGen and Esteban et al. to assess their potential causal effects on pneumoconiosis subtypes (asbestosis, silicosis, and inorganic pneumoconiosis) using conventional and summary-data-based MR (SMR). Gene methylation and expression data from Genotype-Tissue Expression and eQTLGen, along with protein level data from deCODE and UK Biobank Pharma Proteomics Project, were examined in relation to pneumoconiosis data from FinnGen. To validate our findings, we assessed self-measured gut flora from a pneumoconiosis cohort and performed fine mapping, drug prediction, molecular docking, and Phenome-Wide Association Studies to explore relevant phenotypes of key genes. Results Three core gut microorganisms were identified: Romboutsia (OR = 0.249) as a protective factor against silicosis, Pasteurellaceae (OR = 3.207) and Haemophilus parainfluenzae (OR = 2.343) as risk factors for inorganic pneumoconiosis. Additionally, mapping and quantitative trait loci analyses revealed that the genes VIM, STX8, and MIF were significantly associated with pneumoconiosis risk. Conclusions This multi-omics study highlights the associations between gut microbiota and key genes (VIM, STX8, MIF) with pneumoconiosis, offering insights into potential therapeutic targets and personalized treatment strategies. -
Key words:
- Gut microbiota /
- Quantitative trait loci /
- Mendelian randomization /
- Multi-omics
The authors declare no relevant conflicts of interest related to this study.
This research was approved by the First Hospital of Shanxi Medical University Ethics Committee (Approval Number: 2020-k-k104). Before sample collection and clinical data acquisition, written informed consent was obtained from all participants.
The data reported in this study have been deposited at the Beijing Institute of Genomics, Chinese Academy of Sciences. The raw sequence data are available in the Genome Sequence Archive (GSA) at the National Genomics Data Center (NGDC) (GSA Accession Number: CRA013357) and can be accessed at: https://ngdc.cncb.ac.cn/gsa. The remaining GWAS and QTL data were obtained from publicly accessible databases and previously published studies. The supplementary materials will be available in www.besjournal.com.
&These authors contributed equally to this work.
注释:1) Authors’ Contributions: 2) Competing Interests: 3) Ethics: 4) Data Sharing: -
Figure 1. Study design for MR-based analysis of multi-omics and pneumoconiosis risk.
SMR, summary-data-based Mendelian randomization; QTL, quantitative trait loci; VTE, Venous Thromboembolism; SNP, single nucleotide polymorphisms; PPH4, posterior probability of H4; PheWAS, phenome-wide association study.
Figure 2. Associations between genetically predicted Gut microbes and pneumoconiosis in Mendelian randomization analysis (A: Esteban et al., B: MiBioGen Consortium).
OR, odds ratio; CI, confidence interval.
Figure 3. Characterization of gut microbiota in self-assessed pneumoconiosis cohort. (A) Top 10 most abundant gut microbial species; (B) Relative abundance differences in microbial species between groups; (C) Top 10 functional classes of gut microbiota; (D) Differences in gut microbial functional pathways.
Figure 4. Forest plot of genetically predicted four overlapping gut microbes associated with pneumoconiosis in Mendelian randomization analysis.
OR, odds ratio; CI, confidence interval.
Figure 5. Statistical analyses of three core gut microbiota taxa. (A) Scatter plot; (B) Leave-one-out (LLO)plot; (C) funnel plot.
Figure 6. Prediction of potential drugs candidates and molecular docking analysis. (A) GGPS1-Zoledronic acid; (B) GGPS1-Minodronic acid; (C) GGPS1-Ibandronate; (D) PPT1-Palmitic Acid; (E) GM2A-Lauric acid; (F) TPK1-Thiamine; (G) VIM-Artenimol.
GGPS1: Geranylgeranyl Diphosphate Synthase 1; PPT1: Palmitoyl-Protein Thioesterase 1; GM2A: GM2 Ganglioside Activator; TPK1: Thiamin Pyrophosphokinase 1; VIM: Vimentin.
Table 1. In Mendelian randomization analyses, the genes of candidate genes predict methylation, expression, and proteins in relation to pneumoconiosis
Outcome Gene Tier mQTL eQTL (eQTLGen) eQTL (GTEx Lung) eQTL (GTEx blood) pQTL (deCODE) pQTL (UKBppp) Probe OR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueOR (95% CI) P
valueAsbestosis STX8 significant 1.383
(1.037–1.844)0.027 1.735
(1.037–2.902)0.036 2.054
(1.046–4.034)0.037 8.663
(1.161–64.631)0.035 4.23
(1.151–15.545)0.03 VIM significant 2.593
(1.508–4.459)0.001 0.191
(0.041–0.887)0.035 GGPS1 potential 1.64
(1.04–2.58)0.033 1.996
(1.033–3.86)0.04 2.036
(1.032–4.02)0.04 4.433
(1.085–18.115)0.038 NT5C3B potential cg13092355 0.87
(0.768–0.973)0.029 1.14
(1.013–1.283)0.029 1.149
(1.011–1.305)0.033 1.141
(1.013–1.285)0.029 1.228
(1.021–1.477)0.029 GSTM4 potential cg05793930 0.614
(0.415–0.814)0.015 0.815
(0.697–0.953)0.011 0.801
(0.678–0.945)0.009 0.855
(0.761–0.96)0.008 0.885
(0.702–1.116)0.301 0.779
(0.432–1.404)0.407 ACADM potential cg05467918 1.287
(1.011–1.563)0.04 0.694
(0.494–0.976)0.036 0.486
(0.241–0.981)0.044 0.576
(0.338–0.981)0.042 0.581
(0.336–1.005)0.052 0.402
(0.168–0.964)0.041 Inorganic pneumoconiosis MIF significant cg10819733 0.556
(0.334–0.778)0.024 4.559
(1.199–17.333)0.026 1.343
(1.036–1.739)0.026 1.412
(1.043–1.913)0.026 0.014
(0.00023–0.816)0.04 0.052
(0.004–0.612)0.019 PPT1 potential cg04560534 4.23
(1.067–7.393)0.04 0.688
(0.493–0.959)0.028 8.131
(1.038–63.727)0.046 0.482
(0.251–0.925)0.028 9.11
(1.774–46.782)0.008 FBN2 potential cg02271895 3.467
(1.282–5.653)0.014 0.637
(0.429–0.945)0.025 0.681
(0.485–0.956)0.026 0.569
(0.346–0.934)0.026 0.351
(0.141–0.879)0.025 Silicosis GM2A potential 0.676
(0.462–0.989)0.044 0.232
(0.054–0.999)0.05 0.688
(0.478–0.99)0.044 0.271
(0.076–0.968)0.044 1.229
(0.803–1.881)0.341 TPK1 potential cg21370481 1.57
(1.131–2.01)0.007 0.49
(0.272–0.886)0.018 0.305
(0.108–0.864)0.025 0.223
(0.062–0.797)0.021 0.389
(0.177–0.852)0.018 Note. mQTL: Methylation Quantitative Trait Locus; eQTL: Expression Quantitative Trait Locus; pQTL: Protein Quantitative Trait Locus; OR: Odds Ratio; STX8: Syntaxin 8; VIM: Vimentin; GGPS1: Geranylgeranyl Diphosphate Synthase 1; NT5C3B: 5’-Nucleotidase Cytosolic III B; GSTM4: Glutathione S-Transferase Mu 4; ACADM: Acyl-CoA Dehydrogenase Medium Chain; MIF: Macrophage Migration Inhibitory Factor; PPT1: Palmitoyl-Protein Thioesterase 1; FBN2: Fibrillin 2; GM2A: GM2 Ganglioside Activator; TPK1: Thiamine Pyrophosphokinase 1.  下载: 导出CSV
下载: 导出CSV
-
[1] Qi XM, Luo Y, Song MY, et al. Pneumoconiosis: current status and future prospects. Chin Med J, 2021; 134, 898−907. doi: 10.1097/CM9.0000000000001461 [2] Wang DM, Liang RY, Yang M, et al. Incidence and disease burden of coal workers’ pneumoconiosis worldwide, 1990-2019: evidence from the global burden of disease study 2019. Eur Respir J, 2021; 58, 2101669. doi: 10.1183/13993003.01669-2021 [3] Chong SM, Lee KS, Chung MJ, et al. Pneumoconiosis: comparison of imaging and pathologic findings. RadioGraphics, 2006; 26, 59−77. doi: 10.1148/rg.261055070 [4] Adamcakova J, Mokra D. New insights into pathomechanisms and treatment possibilities for lung silicosis. Int J Mol Sci, 2021; 22, 4162. doi: 10.3390/ijms22084162 [5] Gong GC, Song SR, Su J. Pulmonary fibrosis alters gut microbiota and associated metabolites in mice: an integrated 16s and metabolomics analysis. Life Sci, 2021; 264, 118616. doi: 10.1016/j.lfs.2020.118616 [6] Wu YL, Li YH, Luo YB, et al. Gut microbiome and metabolites: the potential key roles in pulmonary fibrosis. Front Microbiol, 2022; 13, 943791. doi: 10.3389/fmicb.2022.943791 [7] Pang JL, Qi XM, Luo Y, et al. Multi-omics study of silicosis reveals the potential therapeutic targets PGD2 and TXA2. Theranostics, 2021; 11, 2381−94. doi: 10.7150/thno.47627 [8] Wang MY, Zhang Z, Liu JF, et al. Gefitinib and fostamatinib target EGFR and SYK to attenuate silicosis: a multi-omics study with drug exploration. Sig Transduct Target Ther, 2022; 7, 157. doi: 10.1038/s41392-022-00959-3 [9] Zhang N, Liu KL, Wang K, et al. Dust induces lung fibrosis through dysregulated DNA methylation. Environ Toxicol, 2019; 34, 728−41. doi: 10.1002/tox.22739 [10] He JY, Zhang CW, Zhang YK, et al. Polyunsaturated fatty acid concentrations and risk of pneumoconiosis: a two-sample mendelian randomization study. Biomed Environ Sci, 2024; 37, 1328−33. [11] Sekula P, Del Greco MF, Pattaro C, et al. Mendelian randomization as an approach to assess causality using observational data. J Am Soc Nephrol, 2016; 27, 3253−65. doi: 10.1681/ASN.2016010098 [12] Zheng J, Baird D, Borges MC, et al. Recent developments in mendelian randomization studies. Curr Epidemiol Rep, 2017; 4, 330−45. doi: 10.1007/s40471-017-0128-6 [13] Lopera-Maya EA, Kurilshikov A, van der Graaf A, et al. Effect of host genetics on the gut microbiome in 7, 738 participants of the dutch microbiome project. Nat Genet, 2022; 54, 143−51. doi: 10.1038/s41588-021-00992-y [14] Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet, 2021; 53, 1712−21. doi: 10.1038/s41588-021-00978-w [15] Sun BB, Chiou J, Traylor M, et al. Plasma proteomic associations with genetics and health in the UK biobank. Nature, 2023; 622, 329−38. doi: 10.1038/s41586-023-06592-6 [16] Võsa U, Claringbould A, Westra HJ, et al. Large-scale cis- and trans-eqtl analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet, 2021; 53, 1300−10. doi: 10.1038/s41588-021-00913-z [17] McRae AF, Marioni RE, Shah S, et al. Identification of 55, 000 replicated DNA methylation QTL. Sci Rep, 2018; 8, 17605. doi: 10.1038/s41598-018-35871-w [18] Kurki MI, Karjalainen J, Palta P, et al. Finngen provides genetic insights from a well-phenotyped isolated population. Nature, 2023; 613, 508−18. doi: 10.1038/s41586-022-05473-8 [19] Chen J, Ruan XX, Sun YH, et al. Multi-omic insight into the molecular networks of mitochondrial dysfunction in the pathogenesis of inflammatory bowel disease. eBioMedicine, 2024; 99, 104934. doi: 10.1016/j.ebiom.2023.104934 [20] Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun, 2017; 8, 1826. doi: 10.1038/s41467-017-01261-5 [21] Legault MA, Perreault LPL, Tardif JC, et al. ExPheWas: a platform for cis-Mendelian randomization and gene-based association scans. Nucleic Acids Res, 2022; 50, W305−11. doi: 10.1093/nar/gkac289 [22] Eberhardt J, Santos-Martins D, Tillack AF, et al. AutoDock Vina 1.2. 0: new docking methods, expanded force field, and python bindings. J Chem Inf Model, 2021; 61, 3891−8. doi: 10.1021/acs.jcim.1c00203 [23] Alikhan MM, Lee FEH. Understanding nontypeable haemophilus influenzae and chronic obstructive pulmonary disease. Curr Opin Pulm Med, 2014; 20, 159−64. doi: 10.1097/MCP.0000000000000023 [24] Mahenthiralingam E. Emerging cystic fibrosis pathogens and the microbiome. Paediatr Respir Rev, 2014; 15, 13−5. [25] Chen SS, Zhang XY, Yang C, et al. Essential role of IL-17 in acute exacerbation of pulmonary fibrosis induced by non-typeable Haemophilus influenzae. Theranostics, 2022; 12, 5125−37. doi: 10.7150/thno.74809 [26] Cai Y, van Putten JPM, Gilbert MS, et al. Galacto-oligosaccharides as an anti-bacterial and anti-invasive agent in lung infections. Biomaterials, 2022; 283, 121461. doi: 10.1016/j.biomaterials.2022.121461 [27] Middleton AM, Dowling RB, Mitchell JL, et al. Haemophilus parainfluenzae infection of respiratory mucosa. Respir Med, 2003; 97, 375−81. doi: 10.1053/rmed.2002.1454 [28] Li QM, Cui Y, Xu BC, et al. Main active components of Jiawei Gegen Qinlian decoction protects against ulcerative colitis under different dietary environments in a gut microbiota-dependent manner. Pharmacol Res, 2021; 170, 105694. doi: 10.1016/j.phrs.2021.105694 [29] Park HJ, Jeong OY, Chun SH, et al. Butyrate improves skin/lung fibrosis and intestinal dysbiosis in bleomycin-induced mouse models. Int J Mol Sci, 2021; 22, 2765. doi: 10.3390/ijms22052765 [30] Li Q, Deng MS, Wang RT, et al. PD-L1 upregulation promotes drug-induced pulmonary fibrosis by inhibiting vimentin degradation. Pharmacol Res, 2023; 187, 106636. doi: 10.1016/j.phrs.2022.106636 [31] Turini S, Bergandi L, Gazzano E, et al. Epithelial to mesenchymal transition in human mesothelial cells exposed to asbestos fibers: role of TGF-β as mediator of malignant mesothelioma development or metastasis via EMT event. Int J Mol Sci, 2019; 20, 150. doi: 10.3390/ijms20010150 [32] Bogdanovic A, Bennett N, Kieffer S, et al. Syntaxin 7, syntaxin 8, vti1 and VAMP7 (vesicle-associated membrane protein 7) form an active SNARE complex for early macropinocytic compartment fusion in Dictyostelium discoideum. Biochem J, 2002; 368, 29−39. doi: 10.1042/bj20020845 [33] Bilan F, Nacfer M, Fresquet F, et al. Endosomal SNARE proteins regulate CFTR activity and trafficking in epithelial cells. Exp Cell Res, 2008; 314, 2199−211. doi: 10.1016/j.yexcr.2008.04.012 [34] Collawn JF, Matalon S. CFTR and lung homeostasis. Am J Physiol Lung Cell Mol Physiol, 2014; 307, L917−23. doi: 10.1152/ajplung.00326.2014 [35] Luo YF, Yi H, Huang XY, et al. Inhibition of macrophage migration inhibitory factor (MIF) as a therapeutic target in bleomycin-induced pulmonary fibrosis rats. Am J Physiol Lung Cell Mol Physiol, 2021; 321, L6−16. doi: 10.1152/ajplung.00288.2020 [36] Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol, 2003; 3, 791−800. doi: 10.1038/nri1200 [37] Calandra T, Bernhagen J, Metz CN, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature, 1995; 377, 68−71. doi: 10.1038/377068a0 [38] Donnelly SC, Haslett C, Reid PT, et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med, 1997; 3, 320−3. doi: 10.1038/nm0397-320 [39] Onodera S, Nishihira J, Iwabuchi K, et al. Macrophage migration inhibitory factor up-regulates matrix metalloproteinase-9 and -13 in rat osteoblasts. Relevance to intracellular signaling pathways. J Biol Chem, 2002; 277, 7865−74. doi: 10.1074/jbc.M106020200 -
 24598+Supplementary Materials.xlsx
24598+Supplementary Materials.xlsx

-

 点击查看大图
点击查看大图
计量
- 文章访问数: 842
- HTML全文浏览量: 348
- PDF下载量: 39
- 被引次数: 0

 Quick Links
Quick Links