

 下载:
下载:

-
Particulate matter (PM) is a heterogeneous mixture of liquid and solid particles suspended in the atmosphere, originating from both natural and anthropogenic sources. PM is particularly harmful because it penetrates deep into the alveoli, entering the systemic circulation, and smaller particles can reach more distant organs and cause widespread physiological effects[1]. Growing evidence indicates that PM also adversely affects the ocular system, an organ continuously exposed to environmental stressors. The retinal pigment epithelium (RPE), a monolayer of pigmented cells situated between photoreceptors and the choroid, plays essential roles in photoreceptor maintenance, phagocytosis of shed photoreceptor outer segments, and regulation of the visual cycle. Due to its high mitochondrial demand and constant exposure to light and oxygen, the RPE is particularly vulnerable to oxidative damage[2]. Recent studies demonstrate that PM exposure increases reactive oxygen species (ROS) generation in human RPE cells, triggering oxidative stress, mitochondrial dysfunction, and inflammatory responses, all of which disturb cellular redox balance and impair energy metabolism[3].
Sustained oxidative and inflammatory stress accelerates cellular senescence in RPE cells, as evidenced by increased senescence-associated β-galactosidase (SA-β-gal) activity, DNA damage, and upregulation of p16INK4a, p21CIP1/WAF1, and senescence-associated secretory phenotype (SASP) factors[4]. Premature senescence has emerged as a pathological contributor to retinal aging and degenerative diseases, including age-related macular degeneration[5]. In this context, premature senescence refers to stress-induced cellular senescence, which occurs independently of replicative aging and is triggered by external stressors such as oxidative stress and mitochondrial dysfunction. However, despite recognition of the harmful ocular effects of PM, the underlying molecular mechanisms driving RPE senescence remain insufficiently defined, and no effective preventive strategy has been established.
Naturally derived bioactive compounds have recently gained attention as potential protective agents against environmental oxidative stress. Among them, compound K (CK), a major triterpenoid saponin metabolite derived from Panax ginseng, has demonstrated potent antioxidant, anti-inflammatory, and cytoprotective properties in multiple cell and tissue models[6]. CK activates the nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) pathway, enhancing endogenous antioxidant defense systems, and modulates AMP-activated protein kinase and sirtuin 1, leading to improved mitochondrial biogenesis and energy homeostasis[7]. Additionally, CK suppresses pro-inflammatory signaling and reduces SASP-related cytokine expression. Given that PM-induced RPE senescence is driven by oxidative stress, mitochondrial dysfunction, and inflammation[3], the pleiotropic activities of CK make it an attractive therapeutic candidate. Therefore, the present study aimed to investigate whether CK can attenuate PM-induced oxidative damage and premature senescence in human RPE cells and to elucidate the underlying molecular mechanisms.
To determine whether PM2.5 induces cellular senescence in RPE cells, ARPE-19 cells (human RPE cell line, American Type Culture Collection, Manassas, MD, USA) were treated with PM2.5 (NIST SRM 1650b; Sigma-Aldrich, St. Louis, MO, USA; particle diameter < 2.5 µm). This material is a standardized diesel particulate matter reference material generated under controlled conditions and is widely used as a representative particulate model in toxicological studies due to its reproducibility and well-characterized composition. The treatment concentration of PM2.5 (20 μg/mL) was determined based on previous studies and preliminary CCK-8 assays assessing cell viability, and was selected to induce a moderate reduction in cell viability (sublethal stress) without causing excessive cell death, thereby allowing mechanistic investigation under controlled conditions. For all experiments, statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test using GraphPad Prism software, and a P-value < 0.05 was considered statistically significant.
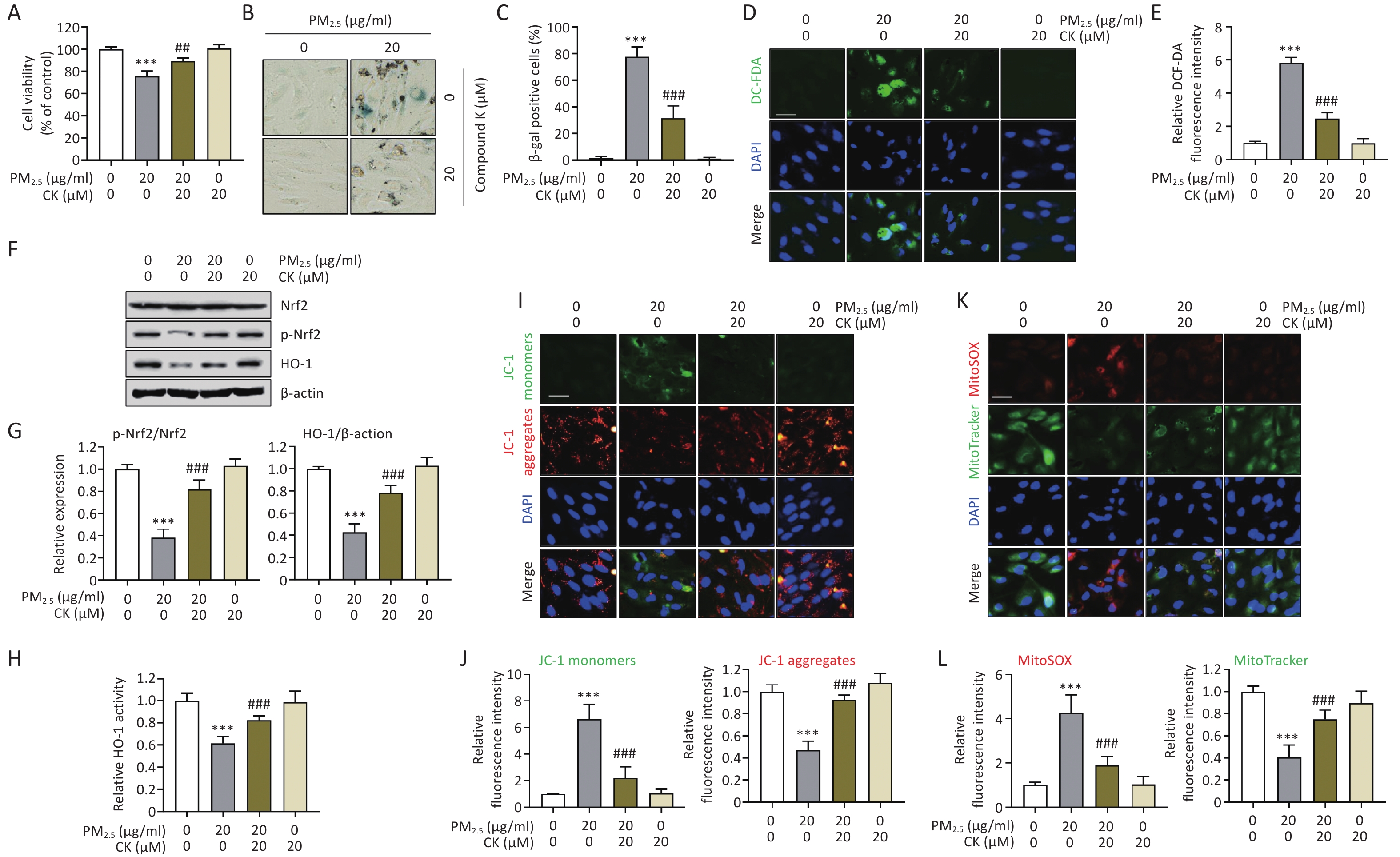
After 24 h of exposure, cell counting kit-8 (CCK-8) and SA-β-gal assays (Sigma-Aldrich) revealed that PM2.5 markedly inhibited cell viability (P < 0.001), suggesting impaired cellular metabolic activity, and increased SA-β-gal activity (P < 0.001, Supplementary Figure S1A-C). Consistent with these findings, 5,6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) staining (Invitrogen, Carlsbad, CA, US) demonstrated that PM2.5 exposure significantly elevated intracellular ROS levels (P < 0.001, Supplementary Figure S1D-E).
Western blot analysis using antibodies against Nrf2 and HO-1 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) showed that PM2.5 decreased Nrf2 phosphorylation and reduced HO-1 expression, without altering total Nrf2 levels (Supplementary Figure S1F and G). Furthermore, HO-1 enzymatic activity measured using a Human HO-1 enzyme-linked immunosorbent assay (ELISA) kit (Abcam Biotechnology, Inc., Cambridge, UK) was also suppressed by PM2.5 treatment (P < 0.001, Supplementary Figure S1H). These results indicate that PM2.5 induces oxidative stress-mediated senescence in RPE cells by suppressing the Nrf2/HO-1 signaling axis. Although ROS is generally known to activate Nrf2 signaling under moderate stress conditions, excessive or sustained oxidative stress may impair this adaptive response[8]. In the present study, the observed increase in ROS accompanied by decreased Nrf2 phosphorylation and HO-1 expression likely reflects a state of oxidative stress overload, leading to dysfunction of the Nrf2 regulatory system. In contrast, CK (20 μM), a concentration selected based on previous studies demonstrating its antioxidant and cytoprotective effects under non-cytotoxic conditions, markedly attenuated PM2.5-induced cellular senescence. Preliminary experiments also confirmed that this concentration did not significantly affect cell viability. CK treatment restored cell viability from 75.79% to 89.37% (P < 0.01), reduced ROS accumulation (P < 0.001), prevented PM2.5-induced Nrf2 dephosphorylation, and recovered HO-1 expression as well as enzymatic activity (P < 0.001, Figure 1A-H).
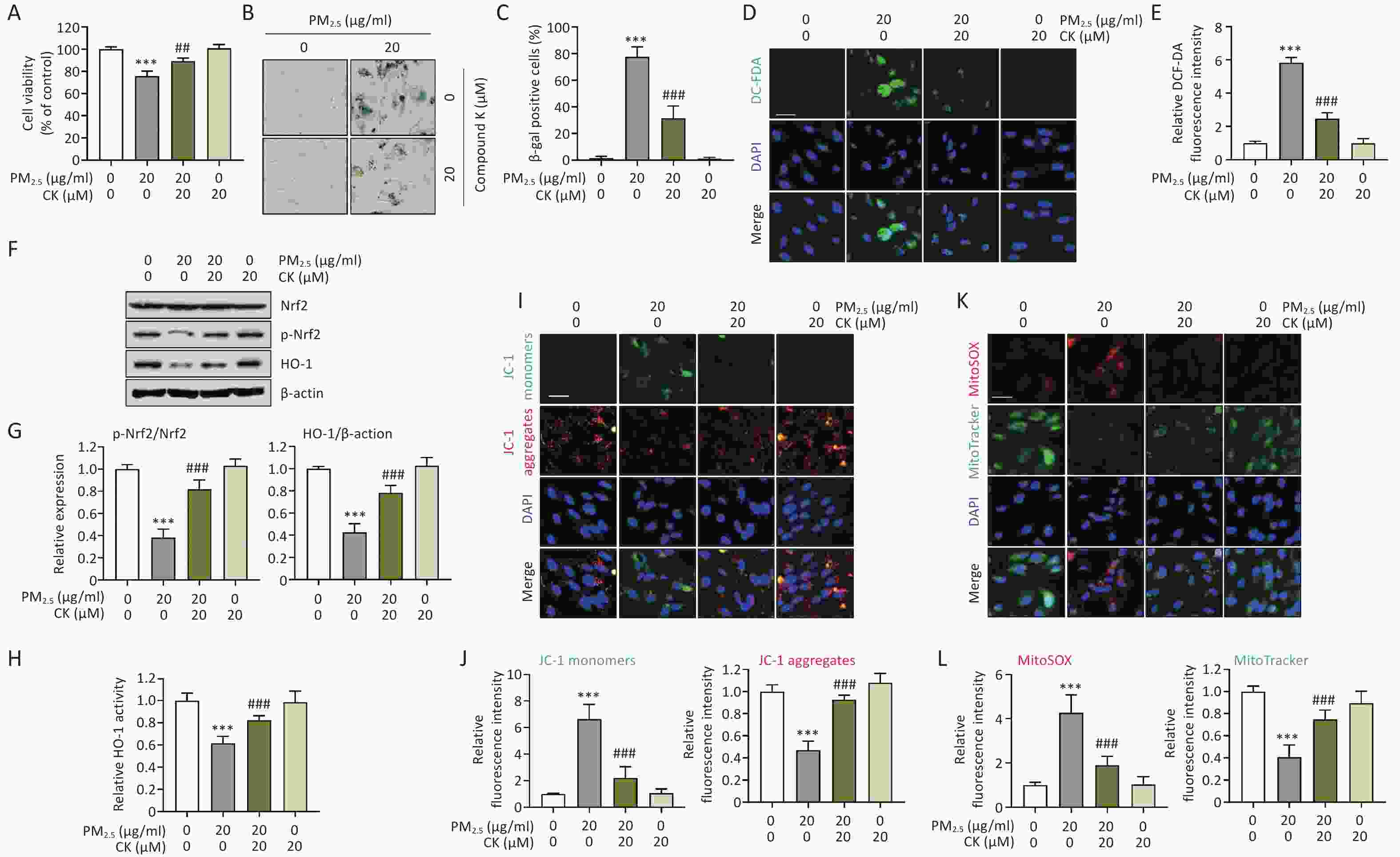
Figure 1. CK restores cytotoxicity, cellular senescence, oxidative stress, inactivation of the Nrf2/HO-1 axis, and impaired mitochondrial homeostasis in ARPE-19 cells induced by PM2.5 exposure. Cells were pretreated with 20 μM CK for 1 h before being treated with 20 μg/mL PM2.5 for 24 h. (A) After treatment, cell viability was measured using the CCK-8 assay. (B and C) Cells were stained for SA-β-gal after PM2.5 exposure, and representative images (B) and quantification of SA-β-gal-positive cells are presented (C). (D and E) To examine intracellular ROS levels, cells were stained with DCF-DA, followed by nuclei staining with DAPI. Representative fluorescence images (D) after staining and the relative values of DCF-DA fluorescence intensity (E) are presented. (F and G) The expression levels of Nrf2, p-Nrf2, and HO-1 were examined by Western blot analysis using total proteins isolated from cells. β-actin was used as a loading control. The protein levels of p-Nrf2 were normalized to total Nrf2 protein levels, and the protein levels of HO-1 were normalized to β-actin, respectively. (H) HO-1 enzyme activity was measured using an ELISA kit. (I and J) To investigate changes in ΔΨm levels, treated cells were stained with JC-1 and then with DAPI, and representative fluorescence images and relative fluorescence intensities of JC-1 monomers and aggregates are presented. (K and L) To assess mitochondrial ROS levels in cells cultured under various conditions, cells were stained with MitoSOX™ Red, a mitochondrial superoxide indicator, followed by MitoTracker, a lipophilic cationic dye that is taken up by the mitochondria. Nuclei were then further stained with DAPI. Representative fluorescence images (J) and relative fluorescence intensities of MitoSOX™ Red and MitoTracker (K) are presented. All results are expressed as mean ± SD. ***P < 0.001 compared to the control group and ##P < 0.01 and ###P < 0.001 compared to PM2.5-treated cells.
To further verify whether PM2.5-induced senescence is linked to mitochondrial dysfunction, 1,1′,3,3′-tetraethyl-5,5′,6,6′-tetrachloroimidacarbocyanine iodide (JC-1) staining (Thermo Fisher Scientific, Waltham, MA, USA) was performed to assess mitochondrial membrane potential (ΔΨm). In ARPE-19 cells exposed to PM2.5, the fluorescence intensity of JC-1 monomers (green), which increases when ΔΨm collapses, was markedly elevated (P < 0.001). In contrast, the intensity of JC-1 aggregates (red), indicative of intact ΔΨm, was significantly reduced (P < 0.001, Figure 1I and J), demonstrating that PM2.5 induces mitochondrial depolarization.
Next, to determine whether the elevated ROS levels originated from mitochondria, cells were stained with MitoSOX™ Red (Thermo Fisher Scientific), a mitochondrial superoxide indicator, together with MitoTracker (Thermo Fisher Scientific), a probe for mitochondrial content and structural integrity. PM2.5 treatment greatly increased MitoSOX™ Red fluorescence (P < 0.001), indicating enhanced mitochondrial superoxide production, whereas the fluorescence intensity of MitoTracker (green) was markedly decreased (P < 0.001), suggesting mitochondrial damage and loss of mitochondrial mass (Figure 1K and L). However, CK pretreatment markedly reversed these mitochondrial alterations (P < 0.001). CK restored ΔΨm, reduced mitochondrial superoxide production, and preserved mitochondrial integrity. These findings suggest that CK protects against PM2.5-induced oxidative mitochondrial damage and maintains mitochondrial homeostasis. Consequently, CK may prevent ROS-mediated cellular senescence by suppressing mitochondrial dysfunction, supporting its potential as a natural therapeutic compound to counteract environmental pollutant-induced retinal aging.
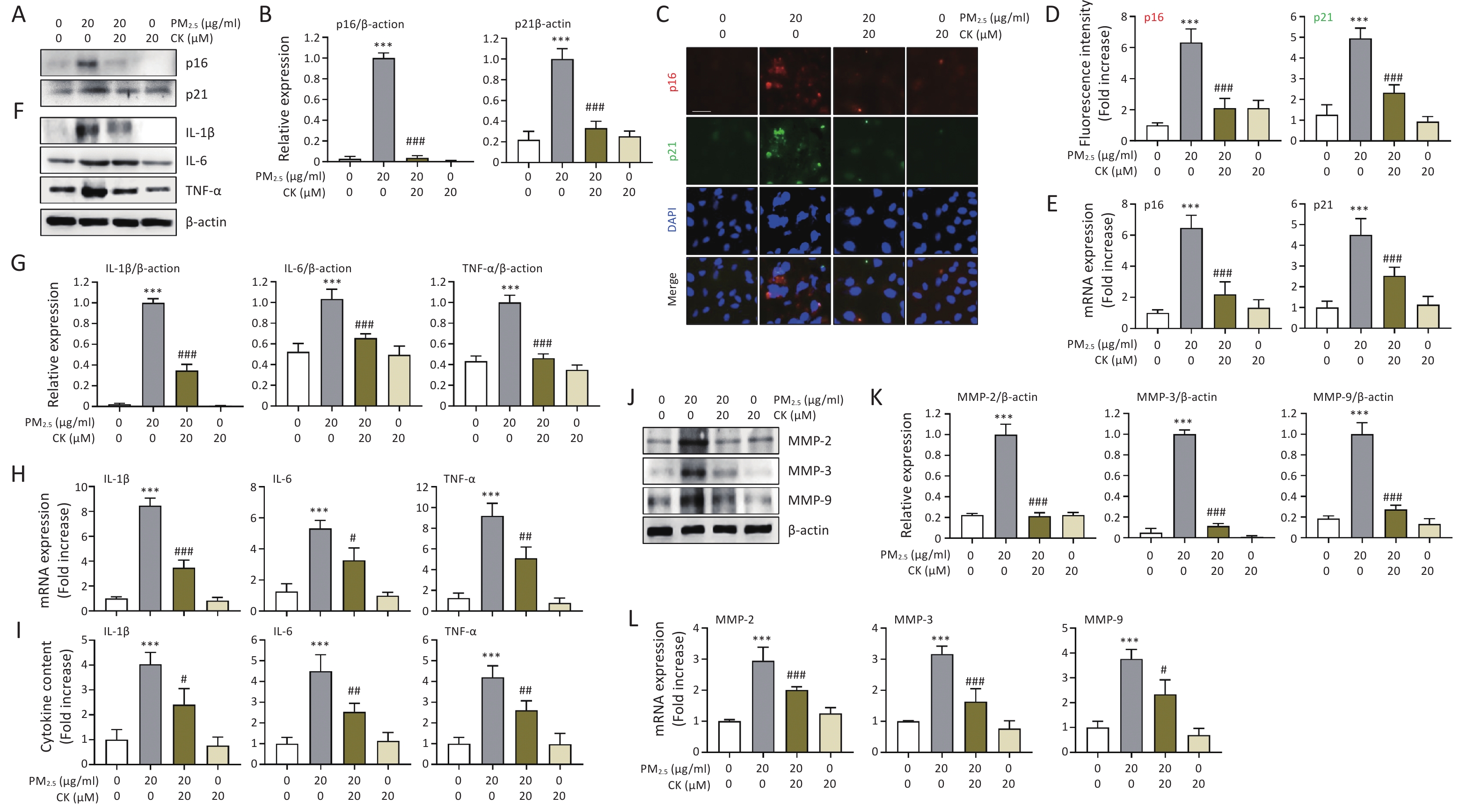
In addition to mitochondrial impairment, we further examined whether PM2.5-induced mitochondrial ROS accumulation translated into activation of the cellular senescence program. Western blot, immunofluorescence, quantitative reverse transcription polymerase chain reaction (qRT-PCR), and ELISA analyses demonstrated that PM2.5 markedly upregulated classical senescence markers, including p16 and p21, at both the protein and mRNA levels (P < 0.001, Figure 2A-E). PM2.5 exposure also significantly increased secretion of SASP factors, such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α, and enhanced matrix metalloproteinase (MMP) activity (P < 0.001, Figure 2F-L), indicating activation of a pro-inflammatory and matrix-degrading senescence phenotype. The concurrent upregulation of p16/p21 and elevation of SASP components following PM2.5 exposure indicate that pollutant-induced mitochondrial dysfunction and ROS accumulation not only arrest the cell cycle but also initiate a deleterious secretory program that propagates inflammation and extracellular matrix remodeling. Notably, CK pretreatment significantly attenuated PM2.5-induced increases in p16 and p21 expression and markedly reduced SASP-related cytokine secretion and MMP activity (P < 0.05-0.001, Figure 2). This reveals a dual protective mechanism of CK: by preserving mitochondrial homeostasis, CK prevents the initiation of the senescence cascade, and by limiting SASP amplification, CK suppresses the paracrine inflammatory and tissue-destructive consequences of senescence. Collectively, these findings indicate that CK exerts anti-senescence activity not merely through antioxidant effects but also by restraining SASP-mediated inflammatory signaling and matrix remodeling, suggesting that CK may serve as a promising natural intervention to prevent or mitigate environmental pollutant-induced retinal aging and inflammation.
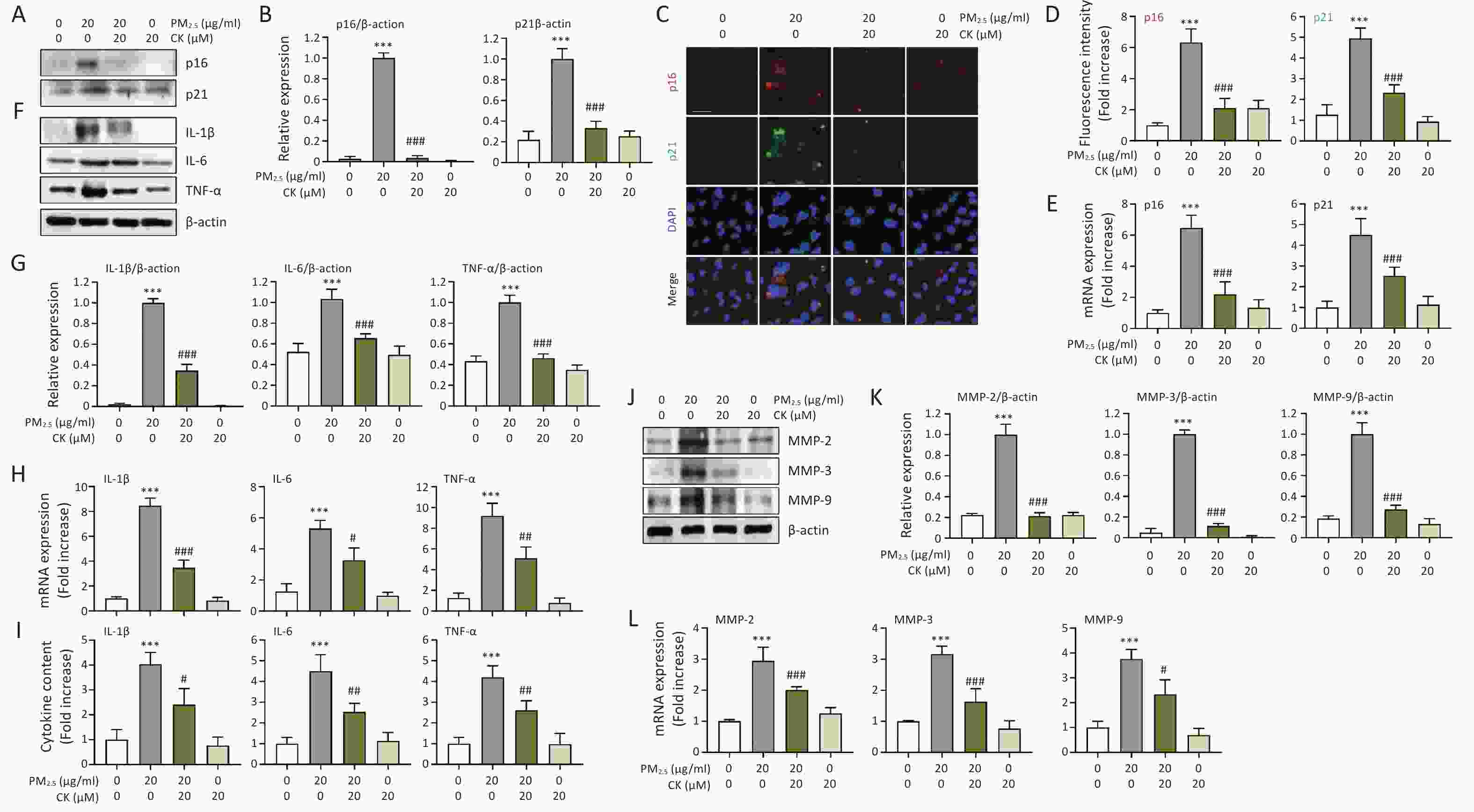
Figure 2. CK suppresses PM2.5-induced senescence markers (p16 and p21) and SASP factors in ARPE-19 cells. Cells were cultured for 1 h in the presence or absence of 20 μM CK were treated with 20 μg/mL PM2.5 and cultured for an additional 24 h. (A, B, F, J and K) The expression levels of the indicated proteins in total cellular protein extracts were examined by Western blot analysis using antibodies against each protein. β-actin was used as a loading control. The levels of each protein were normalized to β-actin. (C and D) Cells were immunostained for the senescence markers (p16 and p21), and DAPI was used to counterstain the nuclei. Representative fluorescence images (C) and quantitative results (D) are presented. (E, G, H and L) The mRNA levels of each gene were measured by qRT-PCR using RNA extracted from cells cultured under different conditions. The relative expression values of each gene were presented relative to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which was used as a housekeeping gene. (I) Protein levels of IL-1β, IL-6, and TNF-α secreted into the medium were quantified using ELISA. All results are expressed as mean ± SD. ***P < 0.001 compared to the control group and #P < 0.05, ##P < 0.01 and ###P < 0.001 compared to PM2.5-treated cells.
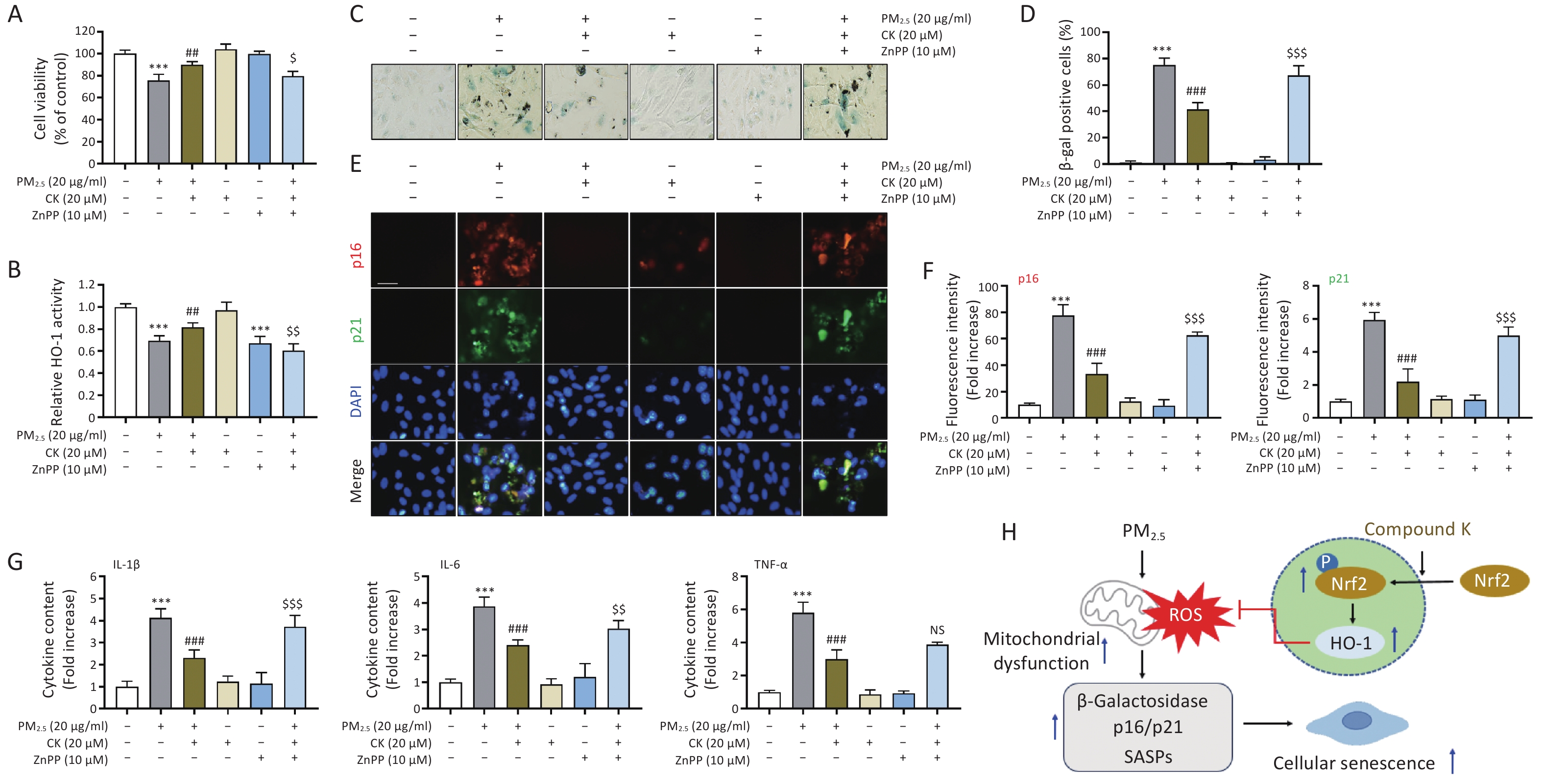
Finally, to verify whether the protective effects of CK against PM2.5-induced cellular senescence are mediated through activation of the Nrf2/HO-1 signaling axis, we used zinc protoporphyrin (ZnPP; Sigma-Aldrich), a competitive inhibitor of HO-1. As shown in Figure 3A, the cytoprotective effect of CK was abolished when HO-1 activity was inhibited by ZnPP (P < 0.05), which corresponded with decreased HO-1 activity (P < 0.01, Figure 3B). Importantly, pharmacological inhibition of HO-1 reversed the anti-senescence effects of CK, as demonstrated by restored SA-β-gal positivity and increased expression of senescence markers (p16 and p21) (P < 0.001, Figure 3C-G). These results suggest that CK functions as a regulator of the Nrf2/HO-1 signaling pathway, enhancing endogenous antioxidant responses. Although ZnPP was used to inhibit HO-1 activity in this study, its potential nonspecific effects should be considered. Therefore, while our findings suggest the involvement of HO-1 in CK-mediated protective effects, further studies using complementary approaches, such as HO-1 activators or genetic modulation, are required to confirm its precise role. In this context, HO-1 appears to contribute to the protective effects of CK against PM2.5-induced senescence, potentially linking mitochondrial protection to the suppression of inflammatory SASP signaling (Figure 3H).
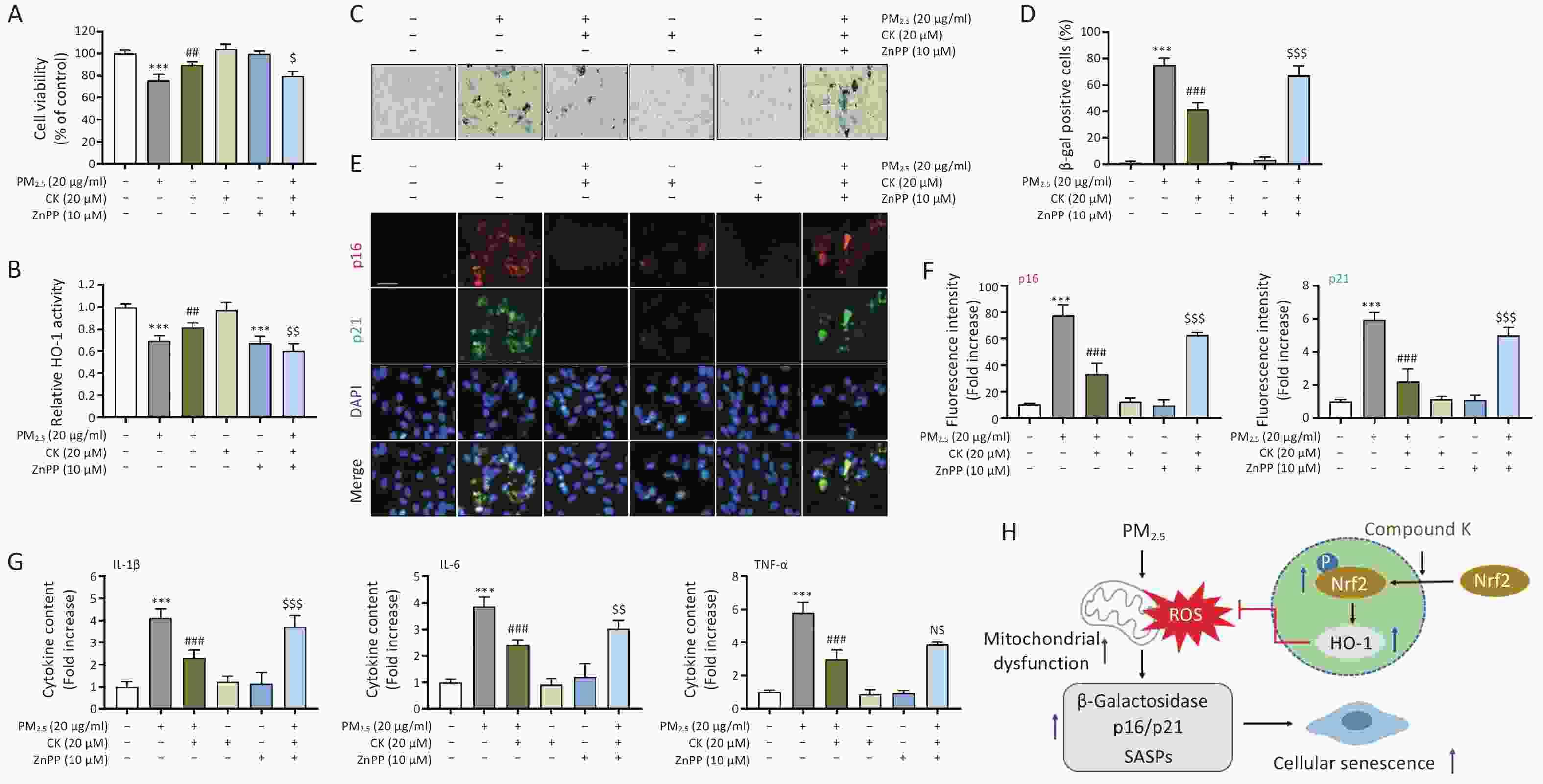
Figure 3. CK protection against PM2.5-induced cellular senescence in ARPE-19 cells requires HO-1 activation. Cells cultured for 1 h in the presence or absence of 20 μM CK and 10 μM ZnPP were treated with 20 μg/mL PM2.5 and cultured for an additional 24 h. (A) Cell viability was measured using the CCK-8 assay. (B) HO-1 enzyme activity was measured using an ELISA kit. (C and D) After SA-β-gal staining, representative images (C) and the quantification results of SA-β-gal positive cells are presented (D). (E and F) Cells were immunostained for the senescence markers (p16 and p21), and DAPI was used to counterstain the nuclei. Representative fluorescence images (E) and quantitative results (F) are presented. (G) Protein levels of IL-1β, IL-6, and TNF-α secreted into the medium were quantified using ELISA. All results are expressed as mean ± SD. ***P < 0.001 compared to the control group, ##P < 0.01 and ###P < 0.001 compared to PM2.5-treated cells, and $P < 0.05, $$P < 0.01 and $$$P < 0.001 compared to PM2.5 and CK-treated cells, NS, not significant. (H) A schematic diagram showing that CK attenuates PM2.5-induced cellular senescence in ARPE-19 cells is presented.
Environmental PM, particularly PM2.5, has emerged as a critical risk factor for ocular surface and retinal disorders[1,3]. In this study, we demonstrate that PM2.5 induces premature senescence in human RPE cells through a mitochondria-driven oxidative stress mechanism and that CK effectively suppresses this process. Our findings reveal a mechanistic cascade in which PM2.5 increases cytosolic and mitochondrial ROS, collapses mitochondrial membrane potential, and disrupts mitochondrial integrity, ultimately activating a p16/p21-dependent senescence program and promoting SASP secretion, including inflammatory cytokines and matrix-degrading MMPs. These events position mitochondrial dysfunction as an upstream trigger that not only arrests the cell cycle but also drives an inflammatory feed-forward loop that may contribute to tissue degeneration. Mitochondrial dysfunction is a critical upstream event that can trigger cellular senescence through excessive production of mitochondrial ROS. Elevated ROS levels can induce oxidative damage to cellular components, including DNA, proteins, and lipids, ultimately activating cell cycle arrest pathways such as p16 and p21. Thus, mitochondrial impairment serves as a key mechanistic link connecting oxidative stress to the initiation of the senescence program. Previous studies, including Park et al.[3], have demonstrated that PM-induced senescence in RPE cells is associated with mitochondrial ROS-mediated downregulation of the Akt/Nrf2 signaling axis, highlighting an upstream regulatory mechanism. In contrast, the present study focuses on the downstream antioxidant response, demonstrating that CK activates the Nrf2/HO-1 pathway and functionally involves HO-1 in the attenuation of senescence and SASP-associated inflammation. Notably, we did not directly assess Akt signaling in the present study. Thus, our findings extend the existing model by providing mechanistic insight into the downstream effector role of HO-1 in regulating PM-induced cellular senescence. In addition to senescence-related mechanisms, previous studies such as Lin et al.[9] have demonstrated that PM2.5 can also induce EMT in ARPE-19 cells through activation of the PI3K/AKT/mTOR pathway. These findings highlight that PM2.5-induced retinal dysfunction involves multiple pathological processes, including both EMT and cellular senescence. While the present study focuses on the senescence axis mediated by oxidative stress and mitochondrial dysfunction, the potential interplay between EMT-related signaling and antioxidant pathways such as Nrf2/HO-1 cannot be excluded. Further studies are required to determine whether these pathways operate independently or converge to regulate PM2.5-induced retinal pathology. It should be noted that the concentration and exposure duration used in this study do not directly reflect physiological in vivo conditions. However, such experimental settings are commonly employed in in vitro studies to induce measurable cellular responses under controlled conditions. Therefore, the findings should be interpreted within the limitations of the in vitro model. In addition, the present study was conducted using a single in vitro model (ARPE-19 cells), and further validation in additional cellular systems or in vivo models will be required to confirm the generalizability of these findings. Although classical senescence markers such as SA-β-gal activity and p16/p21 expression were evaluated, further analysis of additional senescence-associated signaling pathways would provide a more comprehensive understanding and should be addressed in future studies.
A major finding of this work is that the anti-senescence activity of CK is tightly coupled to activation of the Nrf2/HO-1 axis. CK prevented PM2.5-induced mitochondrial depolarization, reduced mitochondrial ROS generation, and preserved mitochondrial morphology, indicating that CK maintains mitochondrial homeostasis under environmental stress (Figure 3). Although the present study demonstrated that CK restored Nrf2 phosphorylation and HO-1 expression, the upstream regulatory mechanisms responsible for Nrf2 activation were not directly examined. Given that Nrf2 activity can be modulated by multiple signaling pathways, including Akt, MAPK, and other stress-responsive kinases, it is possible that CK-mediated protection may involve upstream regulation of these pathways. Further studies are therefore required to elucidate the precise signaling events underlying CK-induced activation of the Nrf2/HO-1 axis.
CK also suppressed SASP amplification, reducing pro-inflammatory cytokines and MMP activity. This finding is particularly meaningful because SASP factors contribute to chronic inflammation and extracellular matrix degradation. The reduction in SASP factors observed in this study is likely an indirect consequence of decreased cellular senescence rather than direct transcriptional suppression. However, the potential involvement of inflammatory signaling pathways, such as NF-κB, cannot be excluded and warrants further investigation. Therefore, CK appears to exert dual protective effects at the cellular level by (1) mitigating the initiation of senescence through preservation of mitochondrial function and (2) limiting SASP-associated inflammatory responses. CK shares common antioxidant mechanisms with other natural Nrf2 activators, such as resveratrol and curcumin[10]. However, as a bioactive ginsenoside metabolite, CK may offer distinct biological relevance, particularly in RPE cells. Notably, the present study extends its functional scope by linking Nrf2/HO-1 activation to mitochondrial dysfunction and cellular senescence. Nevertheless, direct comparative studies are required to determine its relative efficacy and advantages. From a broader perspective, these findings suggest that CK may have potential as a protective agent against PM2.5-induced retinal cellular stress. However, since the present study was conducted in vitro, further in vivo and clinical studies are required to validate its potential therapeutic applicability.
Collectively, our findings suggest that CK may act as a mitochondria-protective and senescence-modulating compound that attenuates PM-induced cellular stress through activation of the Nrf2/HO-1 axis.
doi: 10.3967/bes2026.041
Compound K Mitigates PM2.5-induced Premature Senescence in ARPE-19 Human Retinal Pigment Epithelial Cells through Activation of the Nrf2/HO-1 Pathway
-
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (RS-2025-16064487 and RS-2026-25472831).
The authors declare that they have no competing interests.
This study did not involve animal experiments or human experiments.
Conceptualization: Cheol Park, Cheng-Yun Jin, Gi-Young Kim, and Yung Hyun Choi. Prepared the original draft: Cheol Park, Beom Su Park, and Hyuk Soon Kim Yan. Validation: Beom Su Park, Tae Hwan Shin, Sun-Hee Leem, Jaewon Lee, Heui-Soo Kim, and Cheng-Yun Jin. Methodology: Cheol Park, Beom Su Park, Tae Hwan Shin, Sun-Hee Leem, Hyuk Soon Kim, Jaewon Lee, Heui-Soo Kim, and Gi-Young Kim. Supervised this study: Cheng-Yun Jin, Gi-Young Kim, and Yung Hyun Choi. All authors reviewed the results and approved the final version of the manuscript.
注释:1) Funding: 2) Competing Interests: 3) Ethics: 4) Authors’ Contributions: -
Figure 1. CK restores cytotoxicity, cellular senescence, oxidative stress, inactivation of the Nrf2/HO-1 axis, and impaired mitochondrial homeostasis in ARPE-19 cells induced by PM2.5 exposure. Cells were pretreated with 20 μM CK for 1 h before being treated with 20 μg/mL PM2.5 for 24 h. (A) After treatment, cell viability was measured using the CCK-8 assay. (B and C) Cells were stained for SA-β-gal after PM2.5 exposure, and representative images (B) and quantification of SA-β-gal-positive cells are presented (C). (D and E) To examine intracellular ROS levels, cells were stained with DCF-DA, followed by nuclei staining with DAPI. Representative fluorescence images (D) after staining and the relative values of DCF-DA fluorescence intensity (E) are presented. (F and G) The expression levels of Nrf2, p-Nrf2, and HO-1 were examined by Western blot analysis using total proteins isolated from cells. β-actin was used as a loading control. The protein levels of p-Nrf2 were normalized to total Nrf2 protein levels, and the protein levels of HO-1 were normalized to β-actin, respectively. (H) HO-1 enzyme activity was measured using an ELISA kit. (I and J) To investigate changes in ΔΨm levels, treated cells were stained with JC-1 and then with DAPI, and representative fluorescence images and relative fluorescence intensities of JC-1 monomers and aggregates are presented. (K and L) To assess mitochondrial ROS levels in cells cultured under various conditions, cells were stained with MitoSOX™ Red, a mitochondrial superoxide indicator, followed by MitoTracker, a lipophilic cationic dye that is taken up by the mitochondria. Nuclei were then further stained with DAPI. Representative fluorescence images (J) and relative fluorescence intensities of MitoSOX™ Red and MitoTracker (K) are presented. All results are expressed as mean ± SD. ***P < 0.001 compared to the control group and ##P < 0.01 and ###P < 0.001 compared to PM2.5-treated cells.
Figure 2. CK suppresses PM2.5-induced senescence markers (p16 and p21) and SASP factors in ARPE-19 cells. Cells were cultured for 1 h in the presence or absence of 20 μM CK were treated with 20 μg/mL PM2.5 and cultured for an additional 24 h. (A, B, F, J and K) The expression levels of the indicated proteins in total cellular protein extracts were examined by Western blot analysis using antibodies against each protein. β-actin was used as a loading control. The levels of each protein were normalized to β-actin. (C and D) Cells were immunostained for the senescence markers (p16 and p21), and DAPI was used to counterstain the nuclei. Representative fluorescence images (C) and quantitative results (D) are presented. (E, G, H and L) The mRNA levels of each gene were measured by qRT-PCR using RNA extracted from cells cultured under different conditions. The relative expression values of each gene were presented relative to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which was used as a housekeeping gene. (I) Protein levels of IL-1β, IL-6, and TNF-α secreted into the medium were quantified using ELISA. All results are expressed as mean ± SD. ***P < 0.001 compared to the control group and #P < 0.05, ##P < 0.01 and ###P < 0.001 compared to PM2.5-treated cells.
Figure 3. CK protection against PM2.5-induced cellular senescence in ARPE-19 cells requires HO-1 activation. Cells cultured for 1 h in the presence or absence of 20 μM CK and 10 μM ZnPP were treated with 20 μg/mL PM2.5 and cultured for an additional 24 h. (A) Cell viability was measured using the CCK-8 assay. (B) HO-1 enzyme activity was measured using an ELISA kit. (C and D) After SA-β-gal staining, representative images (C) and the quantification results of SA-β-gal positive cells are presented (D). (E and F) Cells were immunostained for the senescence markers (p16 and p21), and DAPI was used to counterstain the nuclei. Representative fluorescence images (E) and quantitative results (F) are presented. (G) Protein levels of IL-1β, IL-6, and TNF-α secreted into the medium were quantified using ELISA. All results are expressed as mean ± SD. ***P < 0.001 compared to the control group, ##P < 0.01 and ###P < 0.001 compared to PM2.5-treated cells, and $P < 0.05, $$P < 0.01 and $$$P < 0.001 compared to PM2.5 and CK-treated cells, NS, not significant. (H) A schematic diagram showing that CK attenuates PM2.5-induced cellular senescence in ARPE-19 cells is presented.
-
[1] Hamanaka RB, Mutlu GM. Particulate matter air pollution: effects on the respiratory system. J Clin Invest, 2025; 135, e194312. doi: 10.1172/JCI194312 [2] Ridley RB, Amontree AC, Lewin AS, et al. Mitochondrial DNA damage in the retinal pigmented epithelium (RPE) and its role in RPE pathobiology. In: Proceedings of the Mechanisms and Experimental Therapy. Springer. 2025, 375-9. [3] Park BS, Bang E, Hwangbo H, et al. Urban aerosol particulate matter promotes cellular senescence through mitochondrial ROS-mediated Akt/Nrf2 downregulation in human retinal pigment epithelial cells. Free Radical Res, 2024; 58, 841−53. doi: 10.1080/10715762.2024.2438919 [4] Chen YY, Jiang FP, Zeng Y, et al. The role of retinal pigment epithelial senescence and the potential of senotherapeutics in age-related macular degeneration. Surv Ophthalmol, 2025; 70, 942−50. doi: 10.1016/j.survophthal.2025.03.004 [5] Wagh V, Damodaren N, Mittal SK, et al. Cellular senescence: An emerging player in the pathogenesis of AMD. In: Proceedings of the Mechanisms and Experimental Therapy. Springer. 2025, 33-7. [6] Wang XY, Zhang MM, Li YX. Recent studies on the pharmacological activities and structural modifications of compound-K. Mini Rev Med Chem, 2022; 22, 2847−63. doi: 10.2174/1389557522666220513120828 [7] Morshed MN, Akter R, Karim MR, et al. Bioconversion, pharmacokinetics, and therapeutic mechanisms of ginsenoside compound K and its analogues for treating metabolic diseases. Curr Issues Mol Biol, 2024; 46, 2320−42. doi: 10.3390/cimb46030148 [8] Adelusi TI, Du L, Hao M, et al. Keap1/Nrf2/ARE signaling unfolds therapeutic targets for redox imbalanced-mediated diseases and diabetic nephropathy. Biomed Pharmacother, 2020; 123, 109732. doi: 10.1016/j.biopha.2019.109732 [9] Lin HW, Shen TJ, Chen PY, et al. Particulate matter 2.5 exposure induces epithelial-mesenchymal transition via PI3K/AKT/mTOR pathway in human retinal pigment epithelial ARPE-19 cells. Biochem Biophys Res Commun, 2022; 617, 11−7. doi: 10.1016/j.bbrc.2022.05.072 [10] Liu X, Zeng TT, Zhang EF, et al. Plant-based bioactives and oxidative stress in reproduction: anti-inflammatory and metabolic protection mechanisms. Front Nutr, 2025; 12, 1650347. doi: 10.3389/fnut.2025.1650347 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 19
- HTML全文浏览量: 5
- PDF下载量: 0
- 被引次数: 0

 Quick Links
Quick Links