

 下载:
下载:

-
Cervical cancer is a major malignancy that poses a significant threat to women's health[1]. In 2020, an estimated 604,000 new cases and 342,000 deaths were reported globally[2]. The most common pathological subtype is squamous cell carcinoma[3,4]. However, treatment options for advanced cervical squamous cell carcinoma (CSCC) are limited. Surgery is often not feasible at this stage, resulting in poor prognosis[5,6]. Therefore, identifying novel molecular markers and elucidating the mechanisms that drive CSCC growth and metastasis are crucial for improving treatment outcomes.
The Isocitrate Dehydrogenase 2 (IDH2) gene encodes mitochondrial isocitrate dehydrogenase, which is believed to play a role in the development and progression of several malignancies.[7] Studies have shown that IDH2 mutations often occur early in carcinogenesis and influence tumor cell proliferation and metabolism.[8] However, its role of IDH2 in CSCC remains unclear. This study aimed to investigate the function of IDH2 in CSCC and offer new insights into its underlying molecular mechanisms and therapeutic potential.
In this study, we first obtained and processed the RNA sequencing data of 36 cancer types from The Cancer Genome Atlas (TCGA) database to analyze the differences in IDH2 expression among different tumor types. For comparison with normal tissue, we retrieved RNA-seq data from the Genotype-Tissue Expression (GTEx) database. After performing batch correction, we compared IDH2 expression levels in cervical cancer samples from TCGA. Based on this, we divided the patients with CSCC into high-expression and low-expression groups according to the median expression value of IDH2 and compared the clinical characteristics between the two groups. We used the CIBERSORT algorithm to evaluate the correlation between IDH2 expression and immune cell subtypes and analyzed immune cell infiltration in the tumor microenvironment. Following the bioinformatics analysis, we performed in vitro and in vivo experiments. We obtained cervical cancer cell lines (SiHa and C33A), related plasmids and reagents, and nude mice. Cell lines with stable IDH2 knockdown were generated through transfection and validated using quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) and Western Blot analysis. Subsequently, we conducted MTT assay, colony formation, scratch, and cell invasion assays to explore the effects of IDH2 on the proliferation and migration of cervical cancer cells. Next, we constructed a tumor xenograft model, monitored tumor growth, studied the effect of IDH2 on tumor growth in vivo, and analyzed the expression of related proteins using immunohistochemistry (IHC).
To investigate the transcriptional regulation of IDH2, we screened potential transcription factors using JASPAR, CistromeDB, HOCOMOCO, and AnimalTFDB databases. These analyses identified the V-myc avian myelocytomatosis viral oncogene neuroblastoma-derived homolog (MYCN) as a key regulatory factor. Further bioinformatics analysis and luciferase reporter assays confirmed that MYCN directly regulates IDH2 expression. We then constructed MYCN-overexpressing cell lines and evaluated their proliferative and migratory behavior using MTT, colony formation, scratch, and cell invasion assays. Next, to investigate the interaction between MYCN and IDH2, we knocked down IDH2 in MYCN-overexpressing cells and analyzed the resulting effects of the interaction between MYCN and IDH2 on cervical cancer cells using both in vitro and in vivo assays, including IHC. Finally, to explore the downstream mechanism of the MYCN/IDH2 signaling pathway, we conducted Western blot analysis of C33A cells, SiHa cells, and xenograft tumor tissues.
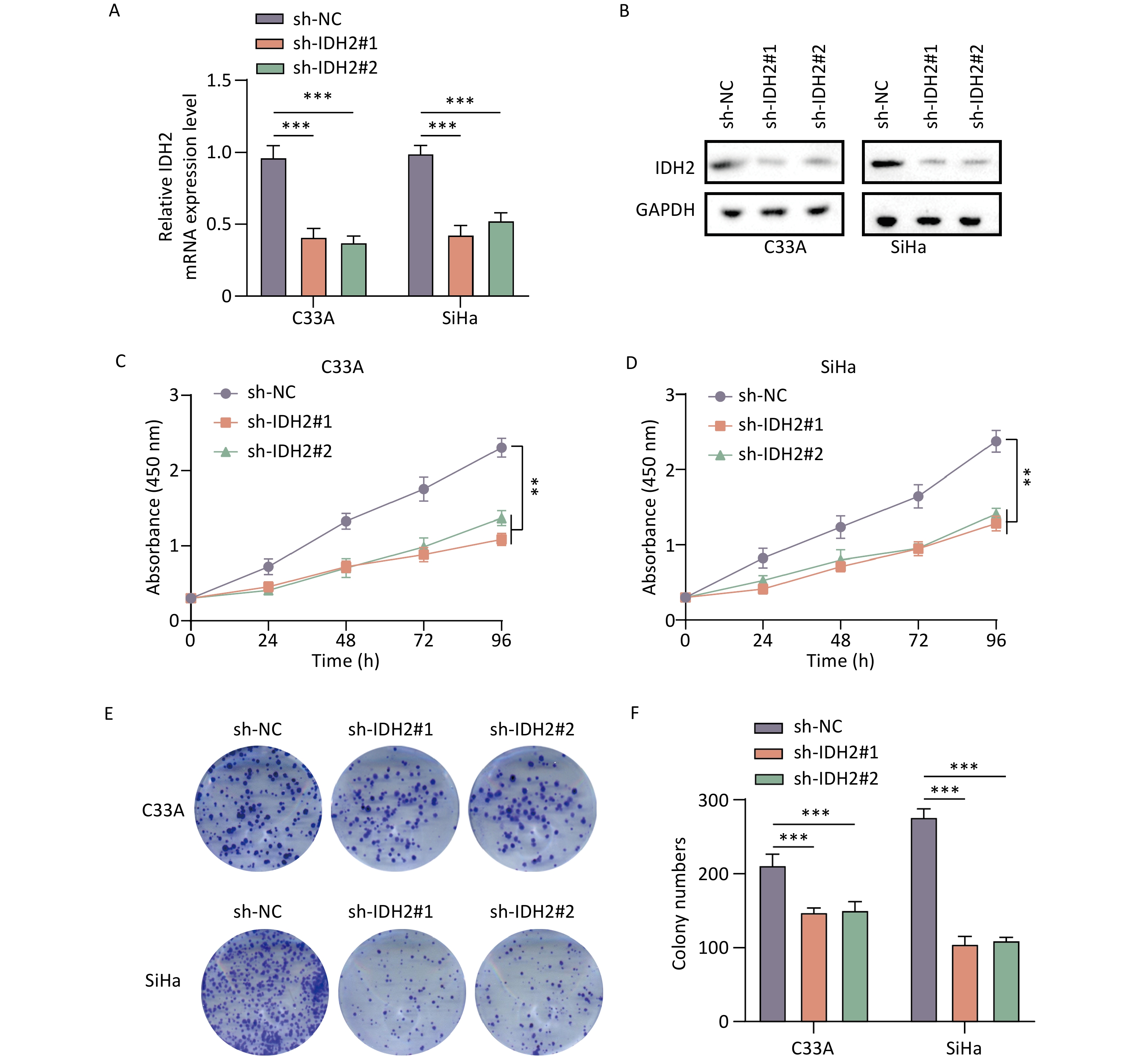
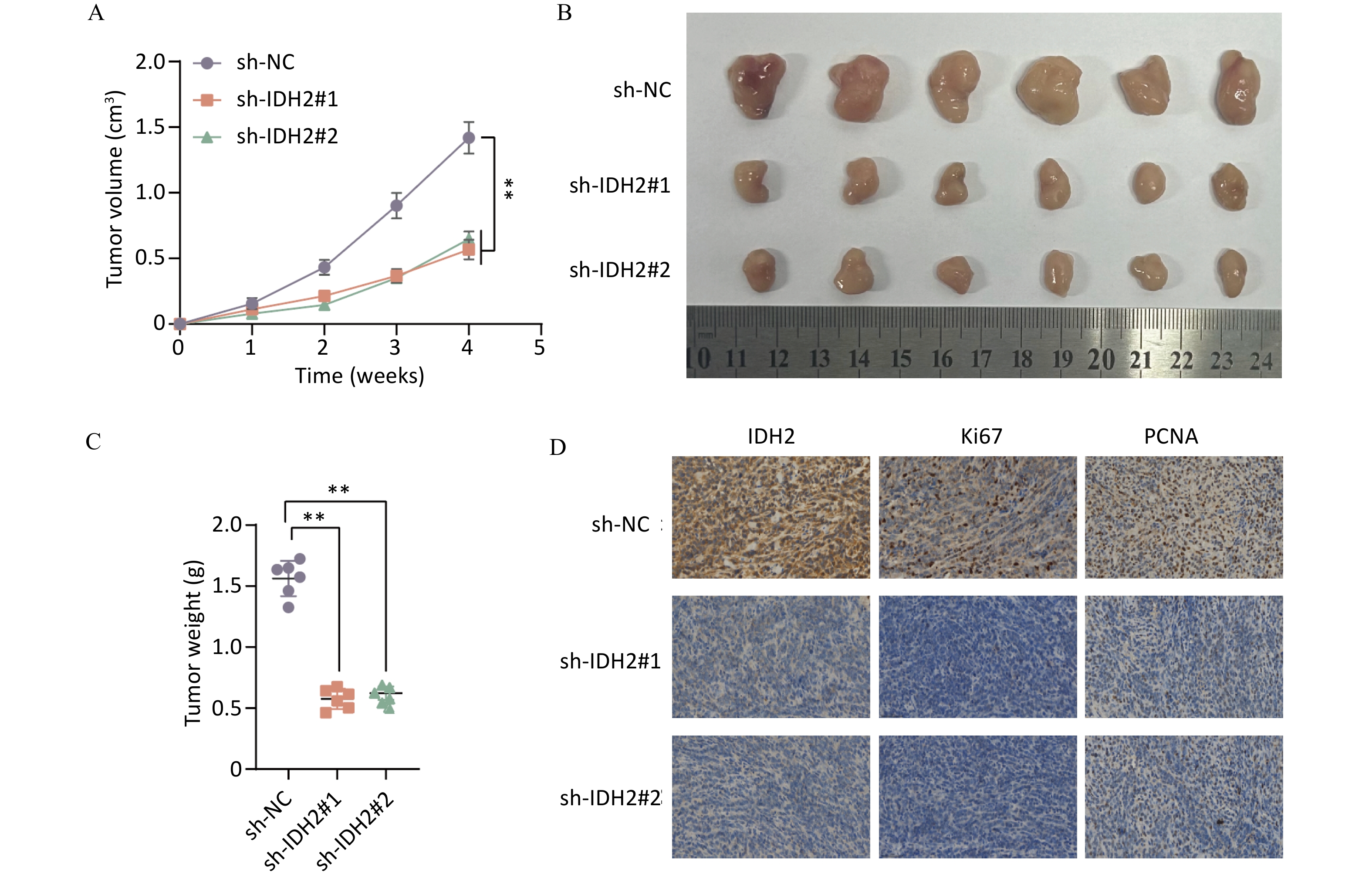
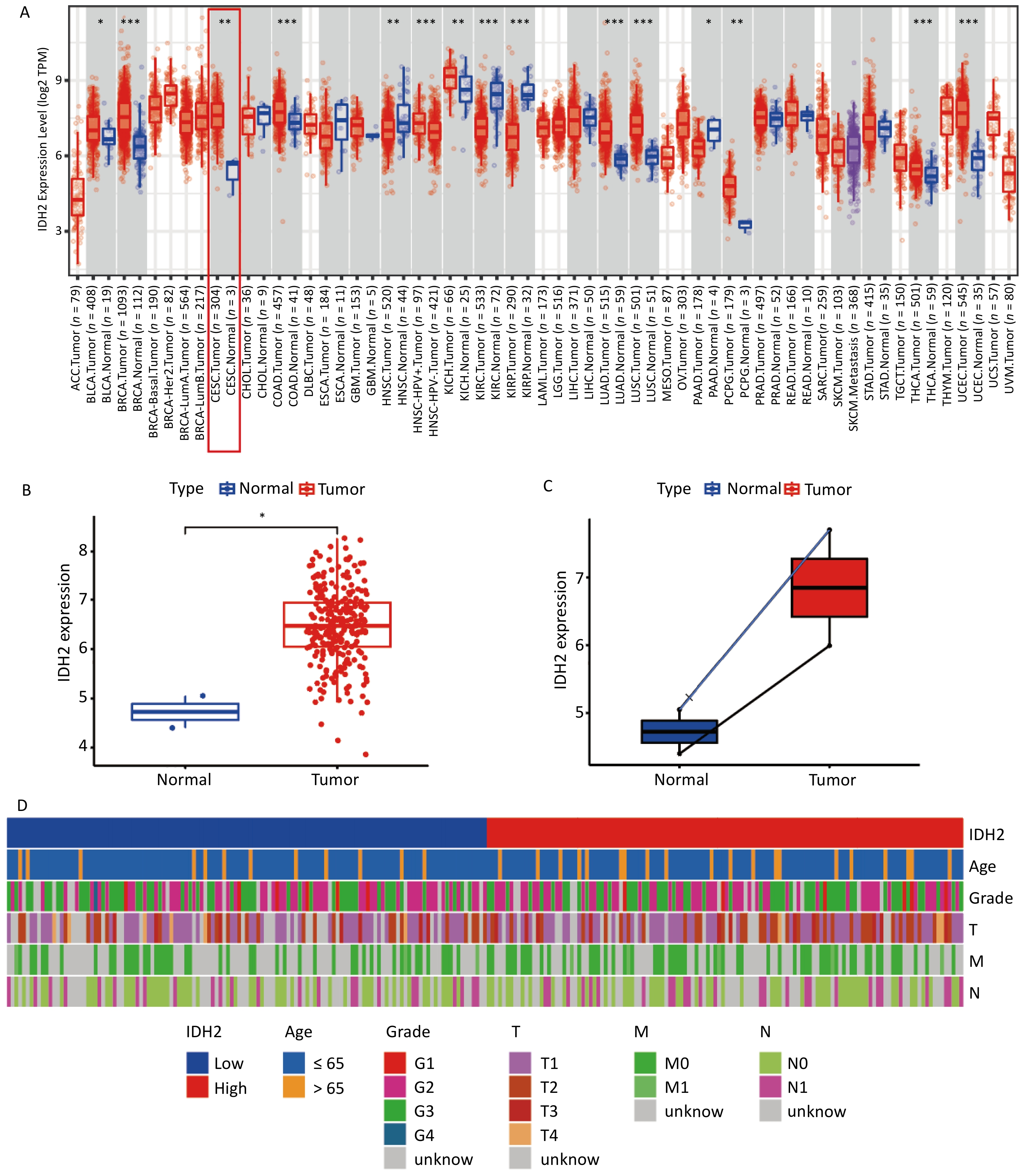
IDH2 is highly expressed in CSCC tissues and correlates with immune cell infiltration. By comparing TCGA and GTEx databases, we found that IDH2 expression was significantly higher in most cancer tissues than in adjacent normal tissues, and its expression in CSCC tissues was significantly higher than that in normal cervical tissues (P < 0.05) (Figure 1A–C). Heatmap analysis revealed no obvious differences in clinical characteristics between patients with high versus low IDH2 expression (Figure 1D). Next, we examined immune cell proportions and observed significant differences between these two groups in follicular helper T cells, resting mast cells, eosinophils, resting natural killer cells, and activated mast cells (P < 0.05) (Supplementary Figure S1A). Correlation analysis indicated that IDH2 expression was significantly negatively correlated with eosinophils and positively correlated with resting mast cells and follicular helper T cells (P < 0.05) (Supplementary Figure S1B–E). Given the elevated expression of IDH2 in CSCC, we used C33A and SiHa cells with IDH2 knockdown to study its effects on cell migration and proliferation. Stable IDH2-knockdown cell lines were verified by qRT-PCR and Western blot (Figure 2A–B). Proliferation assays showed that the proliferative ability of IDH2-knockdown cells was significantly lower than that of the control group cells, indicating that IDH2 silencing inhibited the growth of cervical cancer cells (P < 0.01) (Figure 2C–D). The colony formation assay further confirmed that IDH2 knockdown inhibited the growth and colony-forming abilities of cervical cancer cells (P < 0.001) (Figure 2E–F). Scratch and Transwell invasion assays showed that IDH2 knockdown significantly reduced the migration ability of C33A and SiHa cells (P < 0.001) (Supplementary Figure S2A–F). We investigated IDH2’s role on tumor growth using a mouse xenograft model. The tumor volume in all groups increased over time. Still, the tumor volume and weight of the IDH2-knockdown groups (sh-IDH2#1 and sh-IDH2#2) were significantly lower than those of the control group, indicating that IDH2 knockdown inhibited tumor growth (P<0.01) (Figure 3A–C). IHC analysis confirmed reduced IDH2 expression in knockdown tumors, along with decreased levels of proliferation markers Ki67 and PCNA (Figure 3D). Collectively, these findings indicate that IDH2 promotes CSCC progression.
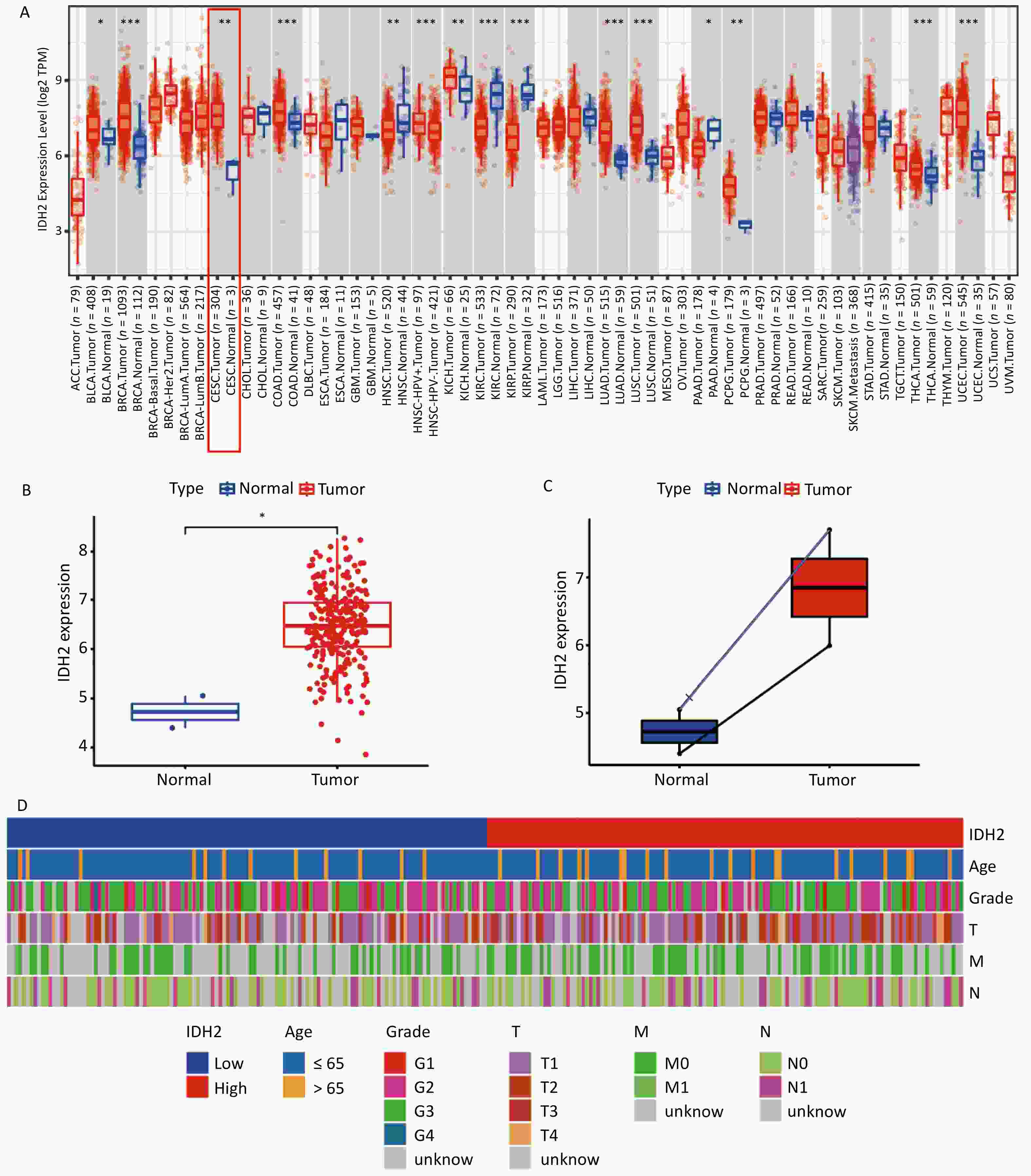
Figure 1. Expression of IDH2 in various cancers (A) Differential expression of IDH2 across cancer types in the TCGA database (B) IDH2 expression in CESC tissues compared to that in normal tissues. (C) IDH2 expression in CESC tissues versus paired adjacent normal tissues (D) Heatmap showing clinical characteristics of patients with high and low IDH2 expression *P < 0.05, **P < 0.01, ***P < 0.001.
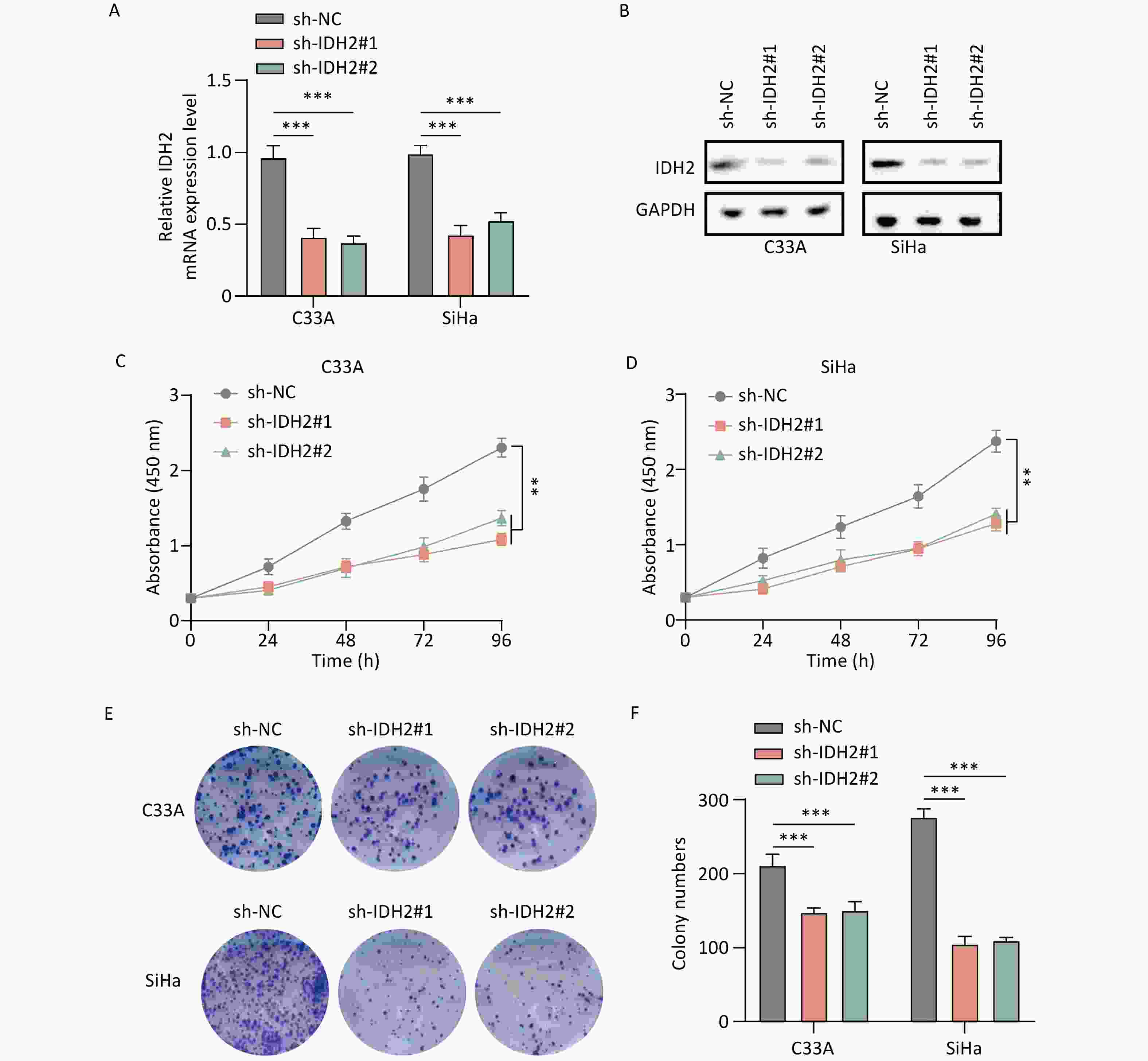
Figure 2. Effect of IDH2 on CESC cell proliferation (A–B) IDH2 mRNA and protein expression in different cell groups (qRT-PCR and WB) (C–D) MTT assays showing reduced proliferation in IDH2 knockdown cells (E–F) Colony formation assays demonstrating suppressed growth in IDH2 knockdown cells **P < 0.01, ***P < 0.001.
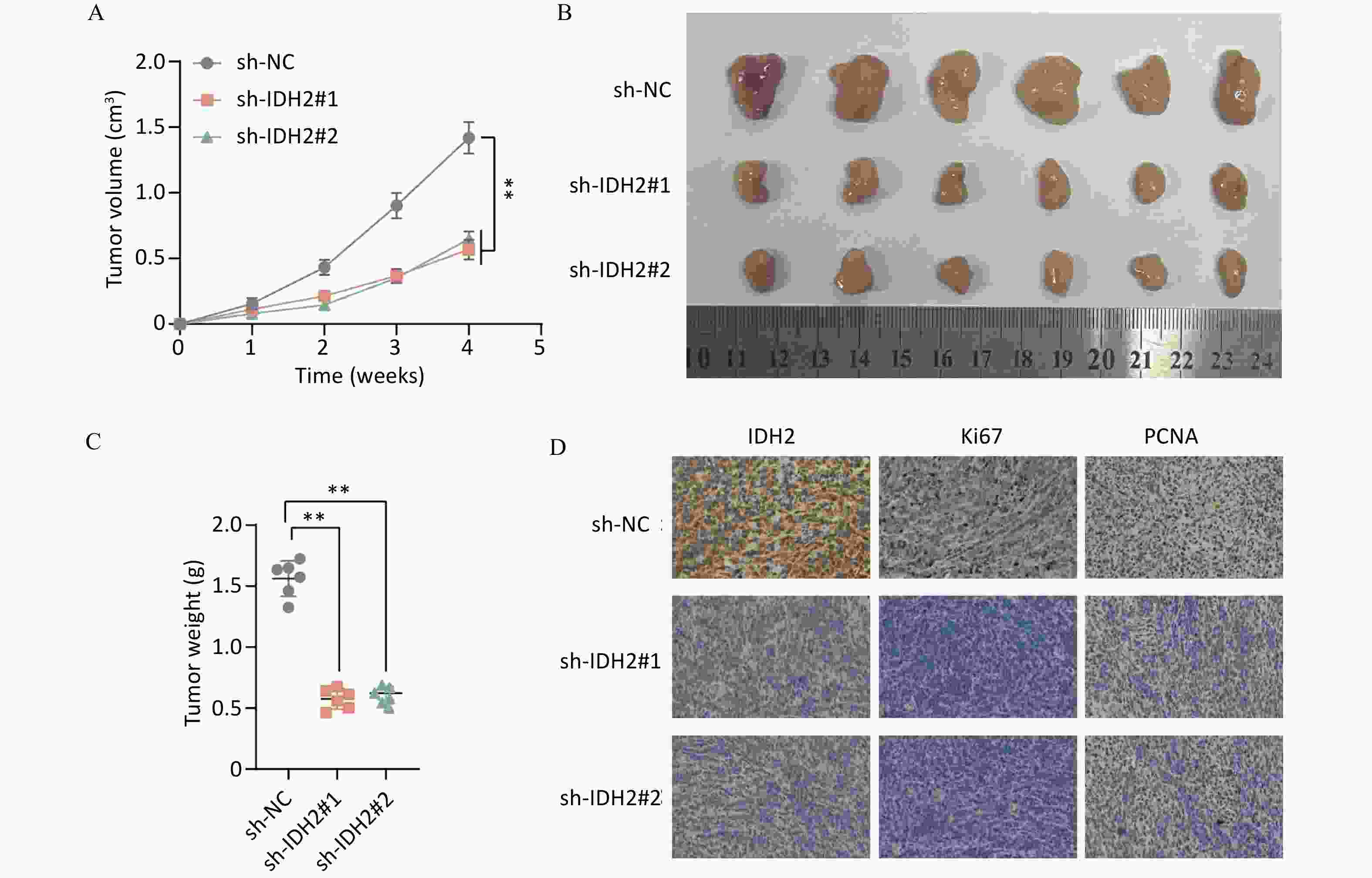
Figure 3. Effect of IDH2 on tumor proliferation in vivo (A–C) Comparison of tumor volume and weight in IDH2 knockdown versus control groups (D) IHC analysis showing reduced IDH2, Ki67, and PCNA expression in tumors from IDH2 knockdown groups **P < 0.01, ***P < 0.001.
To identify transcriptional regulators of IDH2, we queried JASPAR, CistromeDB, HOCOMOCO, and AnimalTFDB. Eight common transcription factors were identified (Supplementary Figure S3A). Integrating this with IDH2 co-expressed genes, we identified MYCN as a key regulator (Supplementary Figure S3B). Bioinformatics showed a positive correlation between MYCN and IDH2 expression (P < 0.05) (Supplementary Figure S3C) and predicted MYCN-binding sites in the IDH2 promoter (Supplementary Figure S3D–E). Luciferase reporter assays confirmed that overexpressing MYCN significantly increased IDH2 promoter activity, while mutation of the primary binding site (PM1) eliminated this effect, indicating that MYCN directly regulated IDH2 expression (Supplementary Figure S3F). We then generated C33A and SiHa cells lines overexpressing MYCN and validated expression levels by qRT-PCR and Western blot (P < 0.001) (Supplementary Figure S4A–B). Proliferation assays showed that MYCN overexpression significantly promoted the growth of cervical cancer cells (Supplementary Figure S4C–D), and the colony formation assay confirmed this result (P < 0.001) (Supplementary Figure S4E–F). Scratch and Transwell assays further indicated that MYCN increased cell migration of C33A and SiHa cells (P<0.001) (Supplementary Figure S5A–F). Additionally, we used a mouse xenograft model to evaluate the effect of MYCN overexpression on tumor growth. The tumor volume and weight in the OE-MYCN group were significantly larger than those in the control group (P < 0.01) (Supplementary Figure S6A–C). IHC analysis revealed elevated levels of MYCN, Ki67, and PCNA in OE-MYCN samples, indicating that MYCN overexpression promoted the proliferation of cervical cancer cells (Supplementary Figure S6D).
To further explore the interaction between MYCN and IDH2, we knocked down IDH2 in MYCN-overexpressing C33A and SiHa cells. In MTT and colony formation assays, MYCN overexpression alone significantly increased proliferation compared to that in controls (P < 0.001). However, cells overexpressing MYCN with concurrent IDH2 knockdown exhibited proliferation rates intermediate between the control and MYCN-only groups (Supplementary Figure S7A–D). Migration analyses, such as Scratch and Transwell assays, further confirmed that the interaction between MYCN and IDH2 regulated cell migration (P < 0.05) (Supplementary Figure S8A–F).
In vivo, tumors derived from the OE-MYCN group were significantly larger and heavier than those from the control group. Although the OE-MYCN+sh-IDH2 group still grew faster than controls, its tumor volume and weight were significantly lower than the OE-MYCN group alone (P < 0.01) (Supplementary Figure S9A–C). These results suggest that MYCN promotes tumor growth mainly by regulating IDH2 expression. IHC on xenograft sections confirmed that MYCN expression elevated IDH2 (Supplementary Figure S9D). Finally, to explore the downstream mechanism of the MYCN/IDH2 signaling pathway, we performed Western Blot analysis on C33A cells, SiHa cells, and xenograft tumor tissues. As shown in Supplementary Figure S10A–B, in cervical cancer cells and tumor models, MYCN overexpression significantly increased the expression levels of IDH2, HIF1-α, and its downstream targets MMP2, MMP9, and VEGF. Conversely, knocking down IDH2 in MYCN-overexpressing cells attenuated the upregulation of HIF1-α, MMP2, MMP9, and VEGF. These results indicate that MYCN promotes tumor proliferation and migration in CSCC by transcriptionally activating IDH2, which in turn drives the HIF1-α signaling pathway.
In conclusion, our integrated bioinformatics analysis, in vitro, and in vivo analyses demonstrate that MYCN directly binds to the IDH2 promoter and drives its transcription in CSCC. IDH2 activation then upregulates HIF-1α, MMP2, MMP9, and EGFR, thereby enhancing tumor proliferation and invasion. This study clarifies the expression patterns, biological functions, and molecular mechanisms of IDH2 in CSCC, highlighting IDH2 as a potential therapeutic target. Future research should further explore the role of the MYCN-IDH2 signaling pathway in the development of CSCC and the therapeutic application of IDH2 inhibition.
doi: 10.3967/bes2025.075
MYCN-Mediated Transcriptional Activation of IDH2 Enhances Proliferation, Migration, and Invasion in Cervical Squamous Cell Carcinoma through the HIF1-α Pathway
-
Conceptualization and funding acquisition: Huiying Zhang. Supervision and funding acquisition: Lisha Shu. Data curation, writing, original draft preparation, and experimental procedures: Xiaojuan Liu. Methodology and Software: Hui Ma. Writing, reviewing, and editing: Xiaoyan Li. software; Validation: Chunxing Ma. All the authors reviewed the results and approved the final version of the manuscript.
The authors declare that they have no competing interests.
This study was conducted in accordance with the principles of the Declaration of Helsinki. This study was approved by the Ethics Committee of the First Affiliated Hospital of Hebei North University (K2024080).
注释:1) Authors’ Contributions: 2) Competing Interests: 3) Ethics: -
Figure 1. Expression of IDH2 in various cancers (A) Differential expression of IDH2 across cancer types in the TCGA database (B) IDH2 expression in CESC tissues compared to that in normal tissues. (C) IDH2 expression in CESC tissues versus paired adjacent normal tissues (D) Heatmap showing clinical characteristics of patients with high and low IDH2 expression *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 2. Effect of IDH2 on CESC cell proliferation (A–B) IDH2 mRNA and protein expression in different cell groups (qRT-PCR and WB) (C–D) MTT assays showing reduced proliferation in IDH2 knockdown cells (E–F) Colony formation assays demonstrating suppressed growth in IDH2 knockdown cells **P < 0.01, ***P < 0.001.
-
[1] Xu MC, Cao CH, Wu P, et al. Advances in cervical cancer: current insights and future directions. Cancer Commun (Lond), 2025; 45, 77−109. doi: 10.1002/cac2.12629 [2] Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin, 2024; 74, 229−63. doi: 10.3322/caac.21834 [3] Cohen PA, Jhingran A, Oaknin A, et al. Cervical cancer. Lancet, 2019; 393, 169−82. doi: 10.1016/S0140-6736(18)32470-X [4] Perkins RB, Wentzensen N, Guido RS, et al. Cervical cancer screening: a review. JAMA, 2023; 330, 547−58. doi: 10.1001/jama.2023.13174 [5] Abu-Rustum NR, Yashar CM, Arend R, et al. NCCN guidelines® insights: cervical cancer, version 1.2024. J Natl Compr Canc Netw, 2023; 21, 1224−33. doi: 10.6004/jnccn.2023.0062 [6] Cibula D, Raspollini MR, Planchamp F, et al. ESGO/ESTRO/ESP Guidelines for the management of patients with cervical cancer - Update 2023. Virchows Arch, 2023; 482, 935−66. doi: 10.1007/s00428-023-03552-3 [7] Klg A, Priyadharshini B, Vasugi S, et al. Exploring the therapeutic potential of biosynthetic enzymes in cancer treatment: Innovations and implications. Int J Biol Macromol, 2025; 292, 139171. doi: 10.1016/j.ijbiomac.2024.139171 [8] Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol, 2021; 18, 645−61. doi: 10.1038/s41571-021-00521-0 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 19
- HTML全文浏览量: 8
- PDF下载量: 1
- 被引次数: 0

 Quick Links
Quick Links