

 下载:
下载:

-
Zearalenone (ZEA), a non-steroidal estrogen-like mycotoxin biosynthesized by several Fusarium species through the polyketide pathway, is also known as the F-2 toxin[1]. ZEA is one kind of common mycotoxins frequently found in feed ingredients and compound feed that can specifically and competitively bind to estrogen receptors resulting in sexual hormone disorder which can negatively affect the expression of secondary sexual characteristics[2]. Sertoli cells (SCs), the somatic cells of the testis, are essential for testis formation and spermatogenesis and are a good model for assessing toxic damage to the male reproductive system in vivo and in vitro. The cell cycle, a series of events that take place in a cell leading to DNA replication and division of cytoplasm and organelles to produce two daughter cells, is divided into four stages: G1, S, G2 and M phase. During cell division, cells develop and establish very precise regulatory mechanisms, and checkpoints that exist in all phases of the cell cycle to identify errors, induce cells to produce specific inhibitors, and impede the progress of the cell cycle[3]. Activation of the 5' adenosine monophosphate-activated protein kinase (AMPK), a regulator of cellular energy balance, is mediated by the ratio of AMP/ATP[4]. Although some recent studies suggest that AMPK can regulate cell proliferation and cell cycle progression, these studies were performed only using tumor cells. For instance, Rattan et al.[5] found that the activation of AMPK by a specific AMPK promoter (AICAR) increases the expression of P53 and other cell cycle-associated proteins, thereby inhibiting the proliferation of various cancer cells. This study was to specifically assess the effect of AMPK pathway activation on the cell cycle distribution induced by Zearalenone (ZEA) in rat Sertoli cells (SCs) in order to elucidate the reproductive toxicity mechanism of ZEA in males and provide a theoretical basis for the prevention and control of ZEA poisoning.
ZEA was purchased from Sigma-Aldrich (St. Louis, MO, USA); Fetal bovine serum (FBS) and DMEM/F-12 medium were obtained from Gibco (Grand Island, NY, USA); RIPA lysate and protease inhibitor complex were purchased from Beijing Pulilai Gene Technology Company (Beijing, China); AMPK inhibitor (Compound C) and AMPK promoter (AICAR) were purchased from MedChem Express (Shanghai, China); Proteins such as P53, P21, Cdc25B, CyclinB1, and Cdc2 were purchased from Santa Cruz (California, USA).
Male Wistar rats of 18–21 d of age and all procedures relating to animal use were approved by the Institute of Comparative Medicine of Yangzhou University. All experimental procedures were performed in accordance with the recommendations of the Care and Use of Laboratory Animals of the National Research Council and were approved by the Animal Care and Use Committee of Yangzhou University [Yangzhou University Medical Center, Approval ID: SYXK (Su) 2017-0044]. SCs were isolated from male Wistar and after the purification of primary Sertoli cells, they were cultured in Dulbecco’s Modified Eagle Medium/F12 supplemented with 10% fetal bovine serum and antibiotics (100 U/mL penicillin G and 100 μg/mL streptomycin) at 37 °C in 5% CO2. ZEA was dissolved in dimethyl sulfoxide (DMSO) and stored at −20 °C.
Flow cytometry analysis was performed using a cell cycle assay kit (BD, 550825) and operated according to the manufacturer’s instructions. The cells were digested with trypsin, centrifuged at 1,000 r/min for 15 min, placed in a 1.5 mL centrifuge tube, fixed in 1 mL 1% paraformaldehyde for 15 min, and stored with pre-cooled 75% ethanol at −20 °C. They were centrifuged at 1,000 r/min for 5 min before analysis. Added 0.5 mL of propidium iodide staining solution to each tube cell sample, and the cells were slowly and fully resuspended, and kept at 37 °C for 30 min in the dark. After staining, a flow cytometer (USA: Beckman Coulter) was used for detection within 24 h, and excited at 488 nm to detect red fluorescence[6].
After the completion of ZEA treatment, the cells were collected using trypsin enzyme-digestion. Total protein from the cells was extracted using RIPA lysis buffer. The concentration of the whole protein was detected and modulated by use of a BCA protein assay kit. The total protein was combined with loading buffer and boiled for 10 min. The extracted proteins were separated on SDS-PAGE according to the size of the protein and then transferred to PVDF membranes (Millipore Corporation, Bedford, MA, USA). The membranes were incubated with 5% skim milk TBST solution then the membranes were probed with the indicated primary antibodies at 4 °C overnight, washed, then incubated with goat anti-rabbit/mouse secondary antibodies for 2 h at room temperature. After being extensively washed with a solution of Tris-buffered saline + Tween 20, they were visualized and analyzed with an ECL detection system which was obtained from Thermo Fisher Scientific (Waltham, MA, USA).
After aspirating the media and washing with PBS, the cells were fixed with 4% paraformaldehyde at 4 °C for 30 min[7]. After rinsing gently with PBS twice, use 0.5% Triton X- 100 to permeate the cell membrane at room temperature for 20 min. After two washes with PBS, the cells were incubated with a 5% BSA sealing solution at room temperature for 20 min. The excess liquid was removed and the cells were incubated with a p-Histone H3 primary antibody solution at 4 °C overnight. After washing with PBS for three times for 2 min each time the following morning, the cells were labeled with Fluorescein isothiocyanate (FITC)-labeled secondary antibody for 1 h. After washing again, the nucleus was stained with Diisopropanolamine (DIPA) for 15 min and the image was observed under a fluorescence microscope[7].
The results were analyzed by using Statistical Product and Service Solutions (SPSS). Results are presented as the mean ± standard deviation (SD). The difference of different groups were analyzed by using a one-way analysis of variance (ANOVA) and means were considered significantly different at P < 0.05.
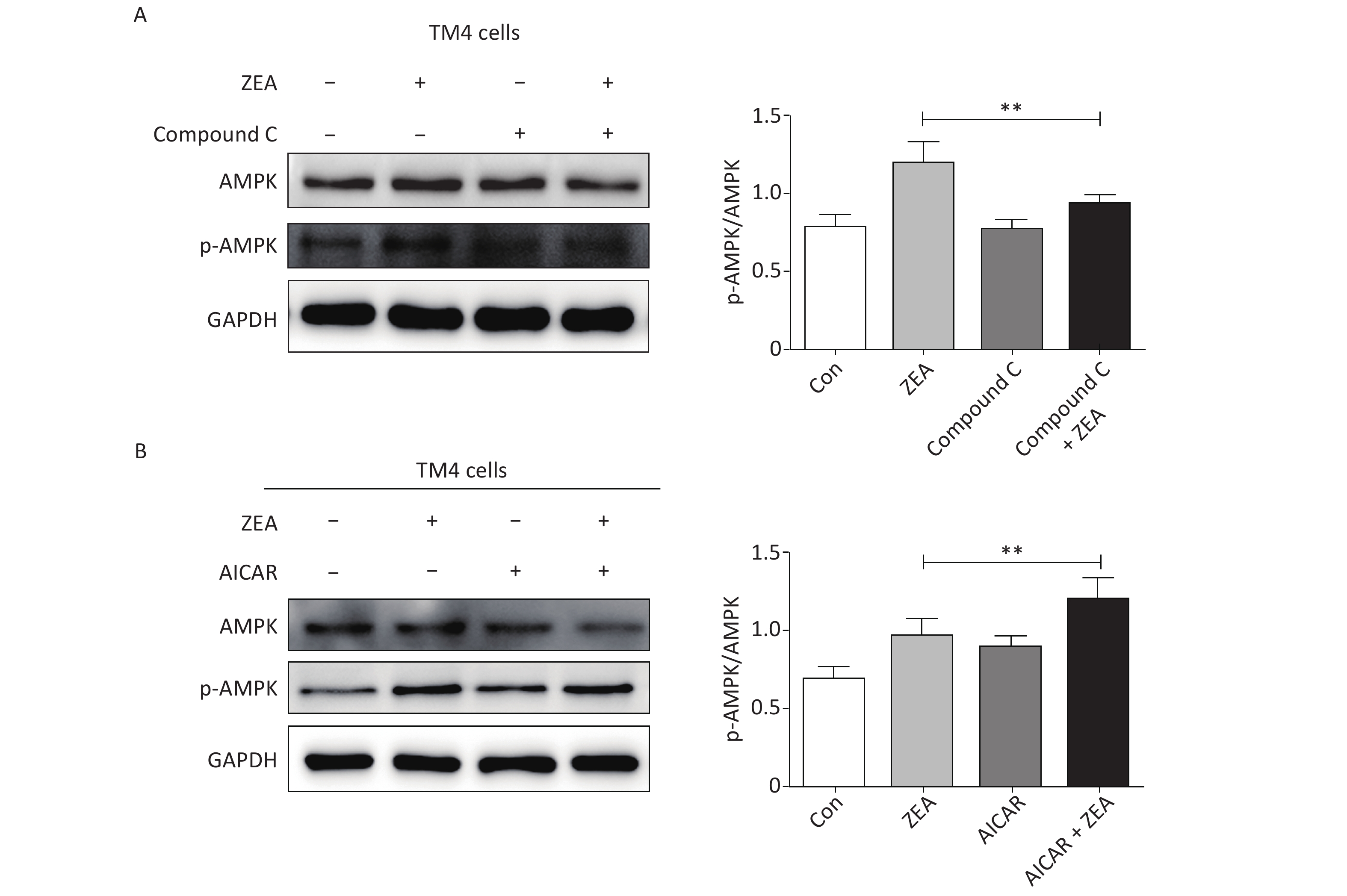
At the beginning, we tested the function of Compound C and AICAR. The log phase SCs were cultured in 100 mm culture dishes, divided into control and treatment groups (20 μmol/L ZEA alone[8], 5 μmol/L AMPK inhibitor (compound C) alone, 5 μmol/L AMPK accelerator (AICAR) alone, co-treatment of 5 μmol/L compound C + 20 μmol/L ZEA, and co-treatment of 5 μmol/L AICAR + 20 μmol/L ZEA), and the expression of AMPK/p-AMPK was examined by western blot. As shown in Figure 1, the expression of AMPK/p-AMPK protein increased in the treatment group 20 μmol/L ZEA alone. Compared to the 20 μmol/L ZEA treatment group, the AMPK/p-AMPK protein expression in the ZEA and Compound C co-treatment group was significantly reduced (P < 0.01) while AICAR co-treatment group was significantly increased (P < 0.01). The data suggested that Compound C and AICAR can effectively change the expression of AMPK and they are fit for AMPK inhibitor and accelerator.
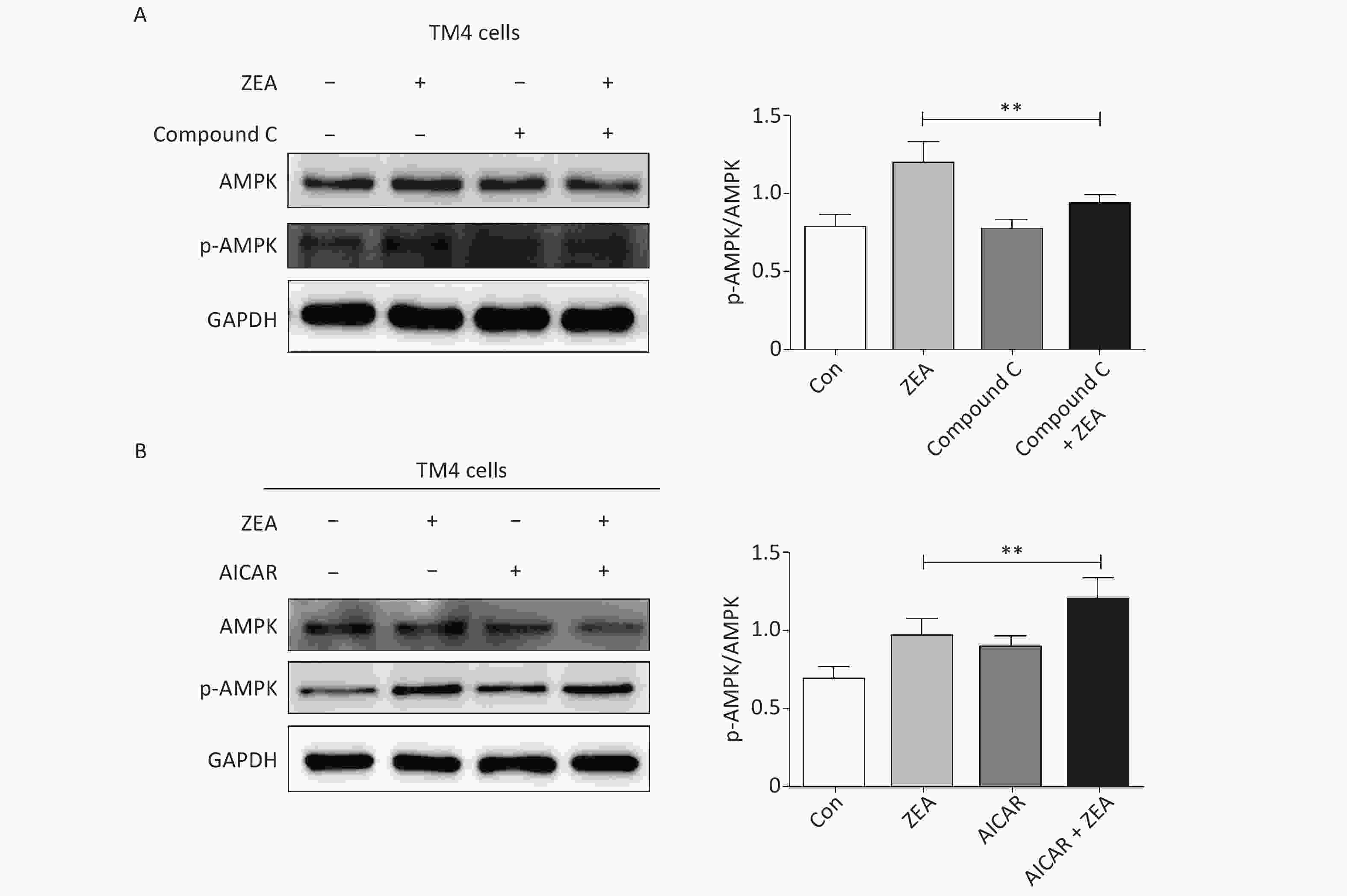
Figure 1. The effects of AMPK pathway promotor and inhibitor on the expression of the AMPK/p-AMPK. (A) The effects of Compound C treatment on the AMPK/p-AMPK expression, **P < 0.01, compared with ZEA alone. (B) The effects of AICAR treatment on the AMPK/p-AMPK expression, **P < 0.01, compared with ZEA alone.
After tested the function of Compound C and AICAR, we used Compound C and AICAR to examine the role of AMPK pathway in ZEA-induced cell-cycle arrest.
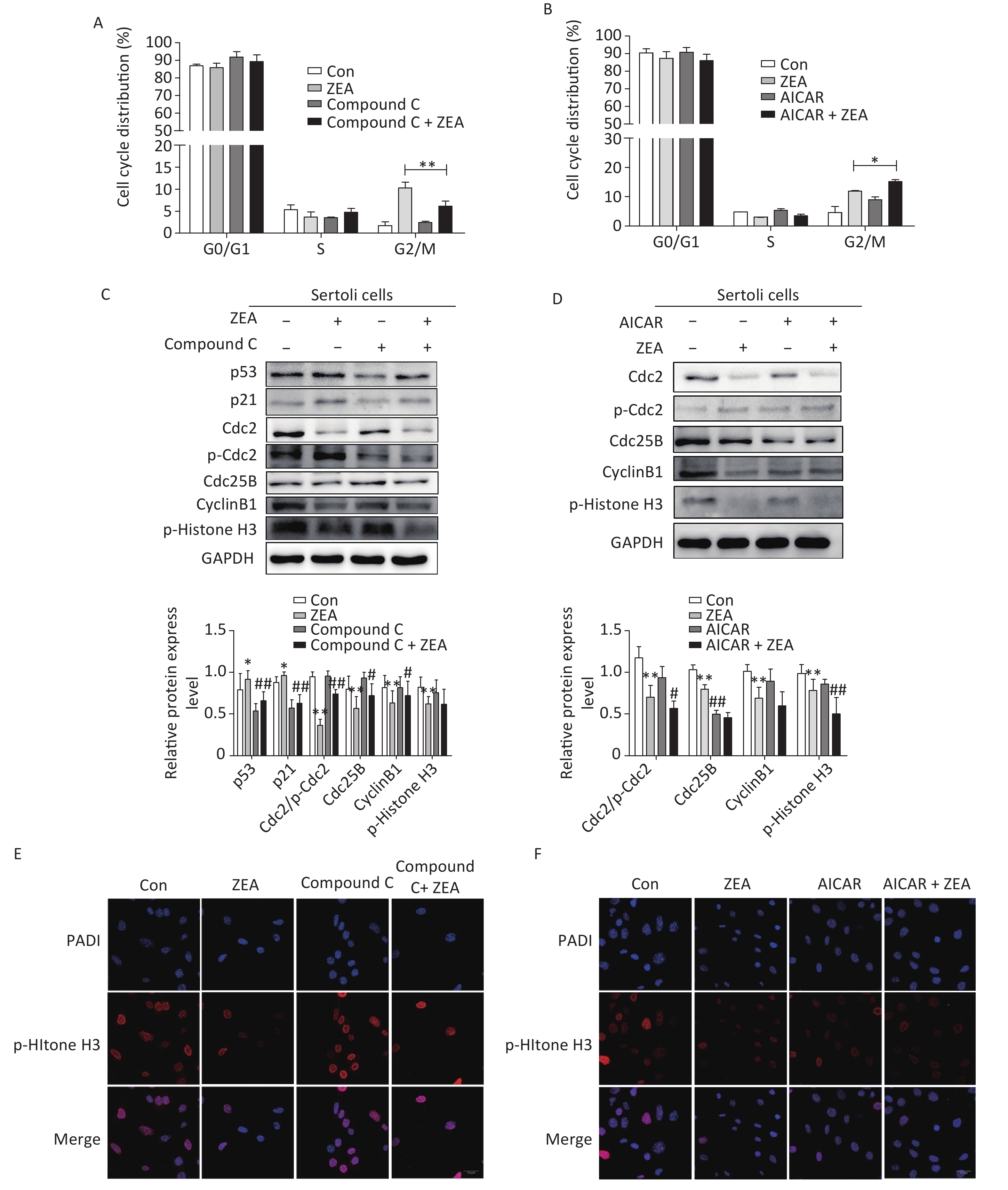
Firstly, to examine the role of Compound C and AICAR on the cell cycle, the distribution of the cell phase was evaluated by flow cytometric analysis when SCs were in the logarithmic growth phase. As shown in Figure 2A and Figure 2B, ZEA induced the arrest of G2/M phases of cell cycle in SCs. Compared to cells treated with ZEA alone, the co-treatment with Compound C significantly alleviated accumulation of the cells in the G2/M phase (P < 0.01), however, the co-treatment with AICAR increased this kind of arrest (P < 0.05). The data suggested that Compound C can partially alleviate ZEA-induced cell cycle arrest, while AICAR can promote this cell cycle arrest.
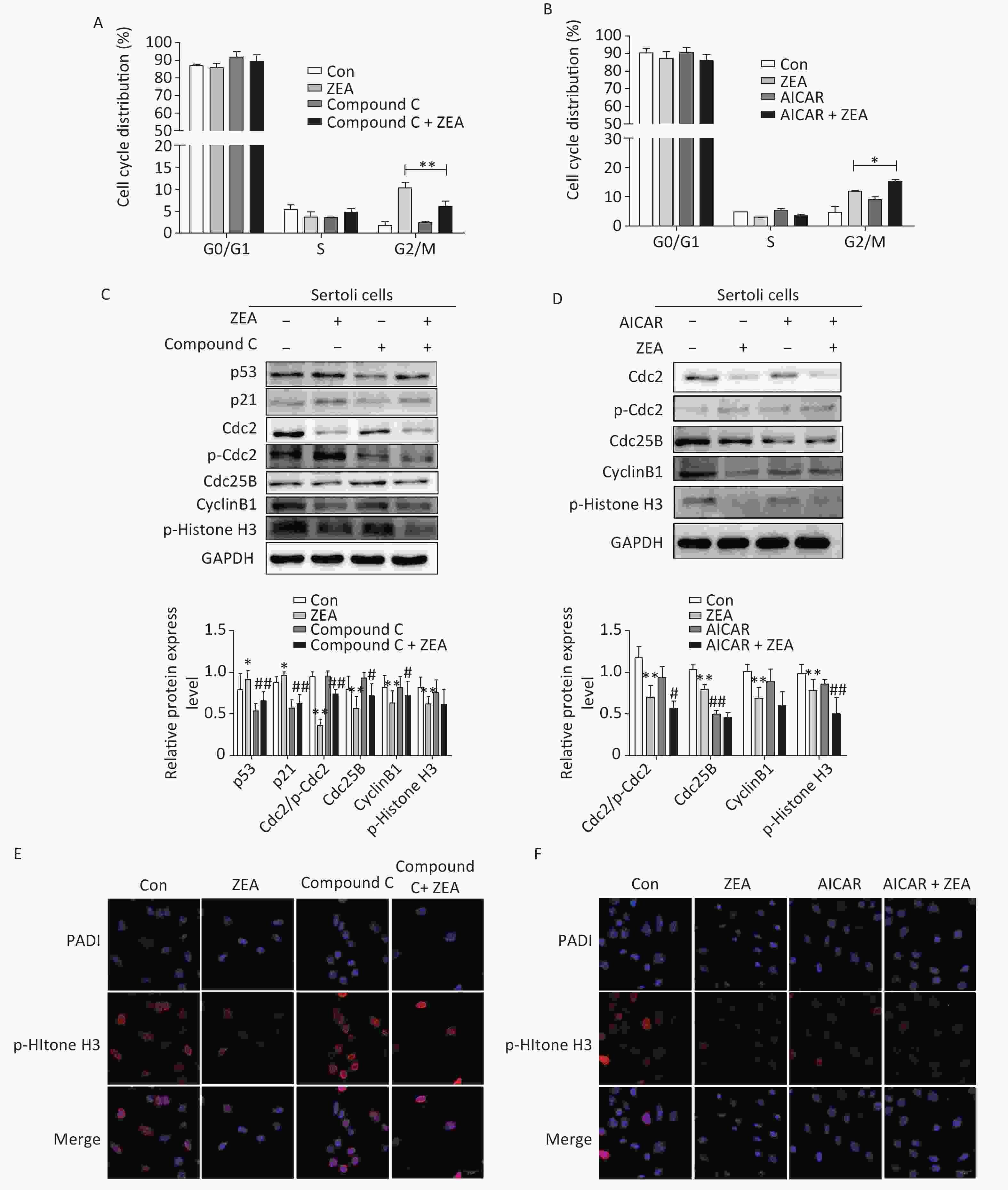
Figure 2. The effects of AMPK pathway on ZEA-induced cell-cycle arrest. (A) The effect of Compound C on cell cycle distribution. **P < 0.01, compared with ZEA alone. (B) The effect of AICAR on cell cycle distribution. *P < 0.05, compared with ZEA alone. (C) The effect of Compound C on ZEA-induced G2 phase regulatory proteins. **P < 0.01, *P < 0.05, compared with control. ##P < 0.01, #P < 0.05, compared with ZEA alone. (D) The effect of AICAR on ZEA-induced G2 phase regulatory proteins. **P < 0.01, *P < 0.05, compared with control. ##P < 0.01, #P < 0.05, compared with ZEA alone. (E) The effect of Compound C on the expression of p-histone H3. (F) The effect of AICAR on the expression of p-histone H3.
Secondly, to elucidate the possible mechanism that Compound C and AICAR contributed different results to the ZEA-induced cell cycle arrest, we evaluated the expression levels of cell cycle-associated regulatory proteins required for G2/M transitions (i.e., Cdc2, Cdc25B, and CyclinB1), proteins (P53 and P21) and the marker protein of the mitotic phase of cells (p-Histone H3) by western blot. The G2/M transition is regulated by CyclinB1-Cdc2 complex which is activated by Cdc25B[9]. Tumor suppressor gene P53 is one of the targets of AMPK that controls cell growth and AMPK activation induces phosphorylation of P53 on serine 15 which is required to initiate AMPK dependent cell-cycle arrest. Expression of the cyclin-dependent kinase inhibitor P21, a downstream mediator of the tumor suppressor P53 in stress conditions, is negatively regulated by the complex activation of CyclinB1/Cdc2[10]. The data (Figure 2C and 2D) indicated that compared to the control group, the protein expression levels of P53 and P21 were increased; in contrast Cdc2/p-Cdc2, Cdc25, CyclinB1 were significantly reduced. Moreover, ZEA also significantly decreased the expression of p-Histone H3 which is consistent with the previous study[8]. In this experiment, co-treatment with Compound C decreased the expression of P51 (P < 0.01) and P21 (P < 0.01), and significantly increased the levels of Cdc2/p-Cdc2 (P < 0.01), Cdc25B (P < 0.05) and CyclinB1 (P < 0.05) (Figure 2C). Once Cdc25B is activated, it causes Cdc2 dephosphorylation, which can enhance the activation of CyclinB1/Cdc2 complex and stimulate G2 conversion in order to reverse the arrest of the G2/M phase. There was no significant effect of the co-treatment group on the expression of p-Histone H3 (P > 0.05) (Figure 2C). But according to the data (Figure 2D), the expression levels of cell cycle regulatory proteins and p-histone H3 were decreased in the group co-treated with AICAR and ZEA. P-histone H3 is a marker protein of the mitotic phase of cells and we will talk about this protein in the next part in detail. Overall the results of western blot indicated blocking the AMPK signaling pathway induces the expression of cycle-associated proteins to alleviate the G2/M phase, however, the acceleration of AMPK activation inhibits these proteins to arrest cells in the G2/M phase.
Thirdly, to further verify the mechanism behind the action of Compound C and AICAR, we evaluated the effects on the expression of phosphorylated histone H3 (p-histone H3) using immunofluorescent staining. The positive expression of FITC-labeled p-histone H3 showed pink fluorescence in the SC nuclei at the mitotic stage, while all nuclei were labeled with DAPI and had blue fluorescence. The results indicated that following treatment with 20 μmol/L ZEA, the expression of p-histone H3 decreased significantly compared to the control group. This indicates that ZEA treatment led to a marked decrease in the proportion of M phase cells and significantly inhibited proliferation of SCs. The results indicated that following the co-treatment with Compound C, the number of cells expressing p-histone H3 were drastically increased compared to the ZEA treatment alone (Figure 2E), however the co-treatment with AICAR showed the opposite results (Figure 2F). The data suggested that the block of AMPK signaling pathway can enhance the key proteins in the M phase to alleviate G2 arrest while activating of the AMPK signaling pathway can enhance G2 arrest by reducing the expression of key proteins in the M phase.
Overall, the results of this study suggested that inhibition of the AMPK pathway can partially reverse the ZEA-induced cell cycle arrest in SCs, and that this arrest can also be augmented by activating the AMPK pathway. Thus, the AMPK pathway plays a significant role in the ZEA-induced arrest in SCs in the G2/M phase. To specifically alleviate this kind of cell cycle arrest, animals should be shielded from certain physiological stressors such as low nutrient intake and prolonged exercise, and also be shielded from pharmacological agents like metformin and AICAR. Nevertheless, additional experiments clearly delineating the involvement of AMPK pathways are warranted.
We appreciate Joshua Flees, Cierla McGuire Sams and Oscar Tejeda for their linguistic assistance during the preparation of this manuscript.
doi: 10.3967/bes2019.120
-
-
Figure 1. The effects of AMPK pathway promotor and inhibitor on the expression of the AMPK/p-AMPK. (A) The effects of Compound C treatment on the AMPK/p-AMPK expression, **P < 0.01, compared with ZEA alone. (B) The effects of AICAR treatment on the AMPK/p-AMPK expression, **P < 0.01, compared with ZEA alone.
Figure 2. The effects of AMPK pathway on ZEA-induced cell-cycle arrest. (A) The effect of Compound C on cell cycle distribution. **P < 0.01, compared with ZEA alone. (B) The effect of AICAR on cell cycle distribution. *P < 0.05, compared with ZEA alone. (C) The effect of Compound C on ZEA-induced G2 phase regulatory proteins. **P < 0.01, *P < 0.05, compared with control. ##P < 0.01, #P < 0.05, compared with ZEA alone. (D) The effect of AICAR on ZEA-induced G2 phase regulatory proteins. **P < 0.01, *P < 0.05, compared with control. ##P < 0.01, #P < 0.05, compared with ZEA alone. (E) The effect of Compound C on the expression of p-histone H3. (F) The effect of AICAR on the expression of p-histone H3.
-
[1] Bennett JW, Klich M. Clinical microbiology reviews. Mycotoxins, 2003; 16, 497−516. [2] Lai FN, Ma JY, Liu JC, et al. The influence of N-acetyl-l-cysteine on damage of porcine oocyte exposed to zearalenone in vitro. Toxicol Appl Pharm, 2015; 289, 341−8. [3] Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Gene Dev, 2004; 18, 2699−711. [4] Motoshima H, Goldstein BJ, Igata M, et al. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol, 2006; 574, 63−71. doi: 10.1113/jphysiol.2006.108324 [5] Rattan R, Giri S, Singh AK, et al. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem, 2005; 280, 39582−93. doi: 10.1074/jbc.M507443200 [6] Fathi E FR, Valipour B, Sanaat Z. Cytokines secreted from bone marrow derived mesenchymal stem cells promote apoptosis and change cell cycle distribution of K562 cell line as clinical agent in cell transplantation. PLoS One, 2019; 14, 4−21. [7] Fathi E, Farahzadi R. Zinc sulphate mediates the stimulation of cell proliferation of rat adipose tissue-derived mesenchymal stem cells under high intensity of emf exposure. Biol Trace Elem Res, 2018; 184, 529−35. doi: 10.1007/s12011-017-1199-4 [8] Wang BJ, Zheng WL, Feng NN, et al. The effects of autophagy and pi3k/akt/m-tor signaling pathway on the cell-cycle arrest of rats primary sertoli cells induced by zearalenone. Toxins, 2018; 10, 398−412. doi: 10.3390/toxins10100398 [9] Hoffmann I, Clarke PR, Marcote MJ, et al. Phosphorylation and activation of human cdc25-C by cdc2--cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J, 1993; 12, 53−63. doi: 10.1002/j.1460-2075.1993.tb05631.x [10] O'Reilly MA. Redox activation of p21Cip1/WAF1/Sdi1: a multifunctional regulator of cell survival and death. Antioxid Redox Sign, 2005; 7, 108−18. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1586
- HTML全文浏览量: 508
- PDF下载量: 80
- 被引次数: 0

 Quick Links
Quick Links