

 下载:
下载:

-
The VATER/VACTERL association (OMIM number: 192350), established in 1973, refers to a specific group of congenital malformations and is closely linked to adverse pregnancy outcomes. Prototypes of multiple congenital anomalies include Vertebral anomalies, Anal atresia, Cardiac defects, Tracheo/Esophageal malformations, Renal dysplasia, Limb abnormalities, and single umbilical artery[1-4]. VACTERL association is usually diagnosed when individuals present with at least 3 of three defects. When individuals present 2 with two malformations, they are diagnosed as VACTERL-like[3]. Although patients with a VATER/VACTERL-like association have no risk of mortality, they suffer from several surgery treatments for multiple congenital anomalies in their lives, which severely reduces their quality of life.
The VACTERL association is rare, with an incidence of 1/10,000–1/40,000 live births; however, the incidence rate was probably underestimated because the significant variability of phenotypes interferes with the clinical diagnosis of patients. When focused on the spectrum of VACTERL phenotypes, Putte et al.[5] reported that 26% of all VACTERL cases were identified as VACTERL-like. Due to the significant heterogeneity in the combinations of VACTERL component features, no distinct subtypes have been identified based on specific phenotypic combinations. Among the VACTERL component features, vertebral anomalies (V), anal atresia (A), and tracheoesophageal malformations (TE) are the most common.
However, the cause of this association remains unclear. Most reported VACTERL association cases are sporadic, with an isolated occurrence and a low risk of having an affected relative. However, several reports have demonstrated evidence of a genetic influence on the familial transmission of the VACTERL association[1,3,6]. Solomon et al.[7] reported that approximately 9% of the patients had primary relatives with at least one VACTERL feature. Studying the genetic origin of the VACTERL association will help expand gene profiles and improve the accuracy of prenatal diagnosis.
In this study, we studied a Chinese family in which four of eight members were confirmed to be afflicted by a VACTERL-like association, presenting with kidney and anorectal malformations. Furthermore, both affected individuals possessed identical microdeletions in Xq27.1. Familial co-segregation analysis suggested that Xq27.1 microdeletion could potentially be a causative factor for VACTERL. This new finding strengthens the evidence supporting the role of inheritance in the manifestation of VACTERL and elucidates the potential implications of Xq27.1 microdeletion.
-
Clinical data on general health, medical history, clinical information, and physical examination results were collected during face-to-face interviews. This study was approved by the Ethics Committee of Beijing Hospital. All the participants provided written informed consent. The diagnosis of VACTERL association was established when an individual presented at least three pertinent clinical features; other differential diagnoses were excluded. When only two of the VACTERL component features were present, the condition was classified as VACTERL-like. This distinction ensures a more accurate and specific diagnosis that adheres to established clinical guidelines[2]. Pedigree analysis was conducted at this step (Figure 1). Blood samples for laboratory analyses and DNA extraction were collected during the first appointment.
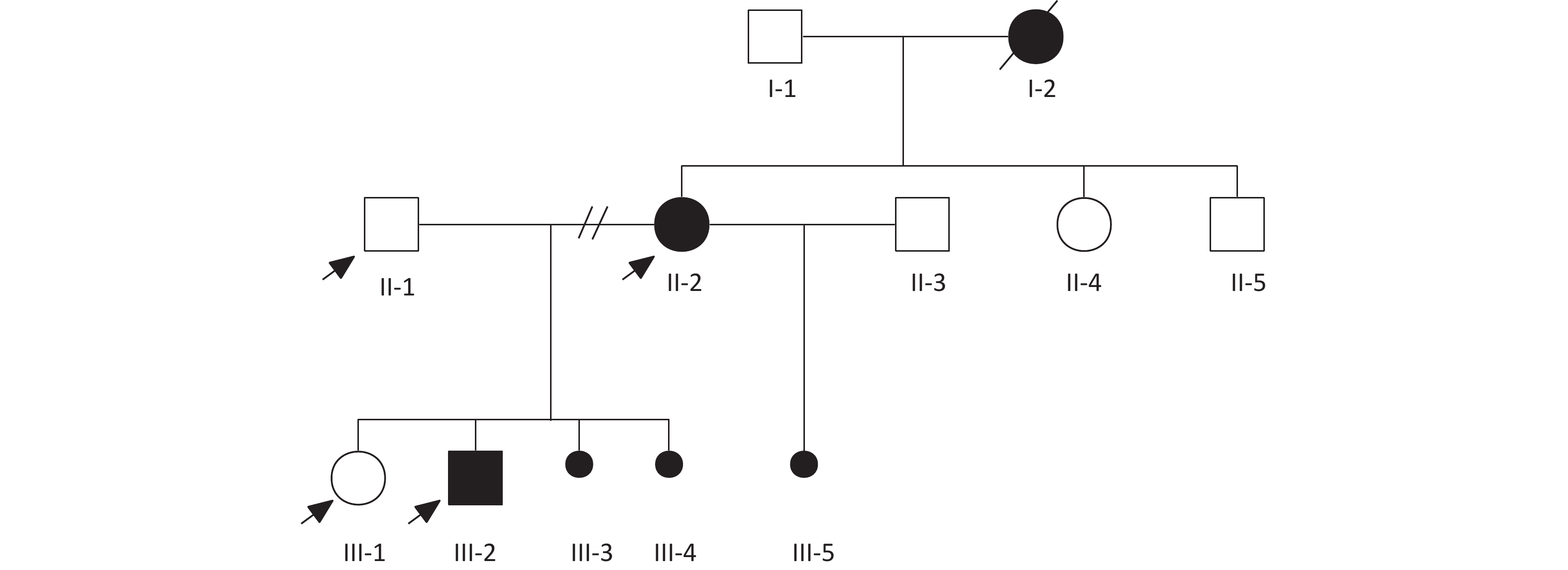
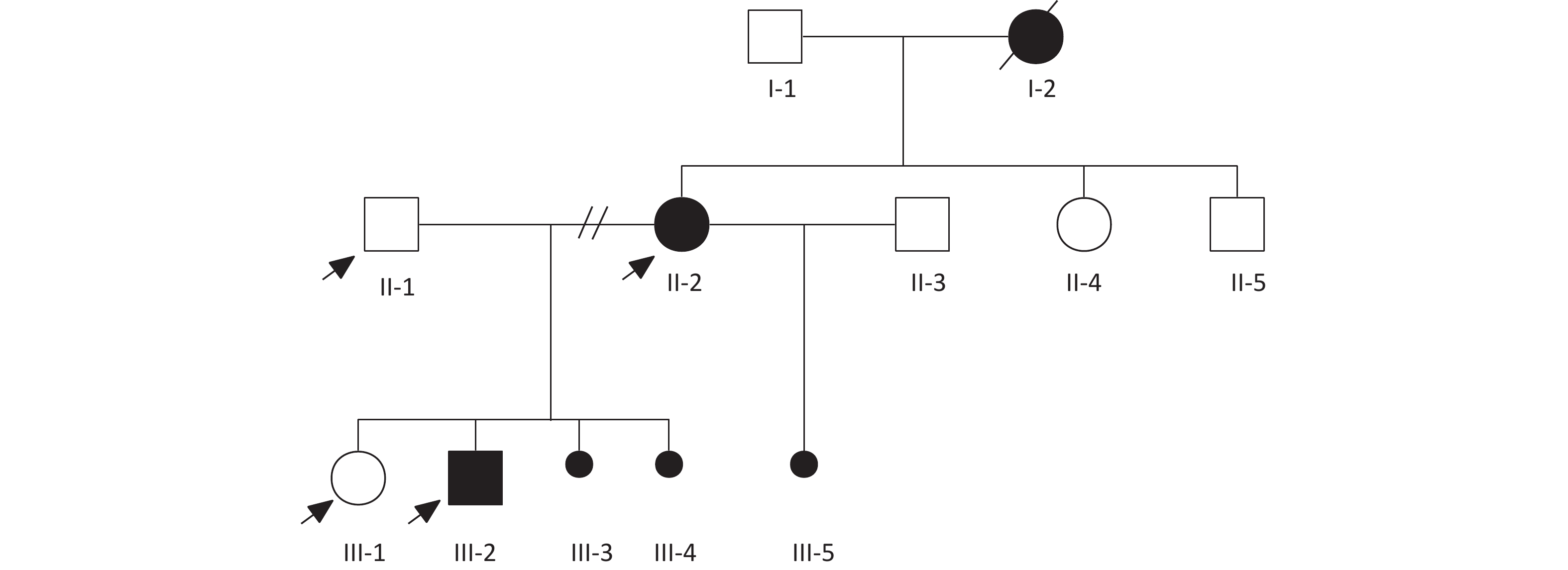
Figure 1. Family pedigree analysis.
-
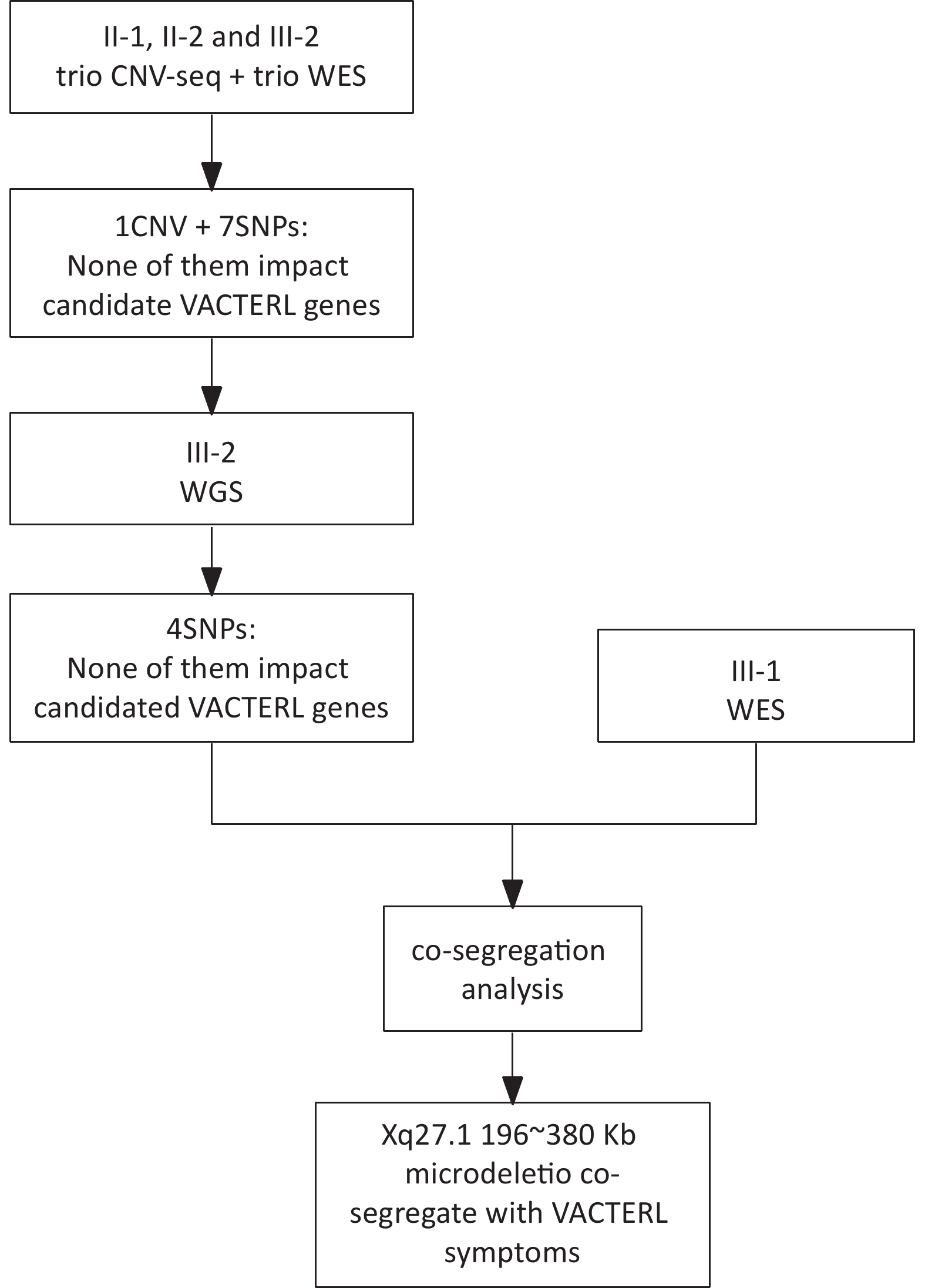
Initial genetic analyses were performed using next-generation sequencing (NGS). Subjects II-1, II-2, and III-2 underwent both copy number-variation sequencing (CNV-seq) and whole-exome-sequencing (WES) for a comprehensive genomic assessment. Subsequently, one male patient (III-2) underwent whole-genome sequencing (WGS) for an in-depth investigation, whereas his older sister underwent WES to assist with co-segregation (Figure 2).
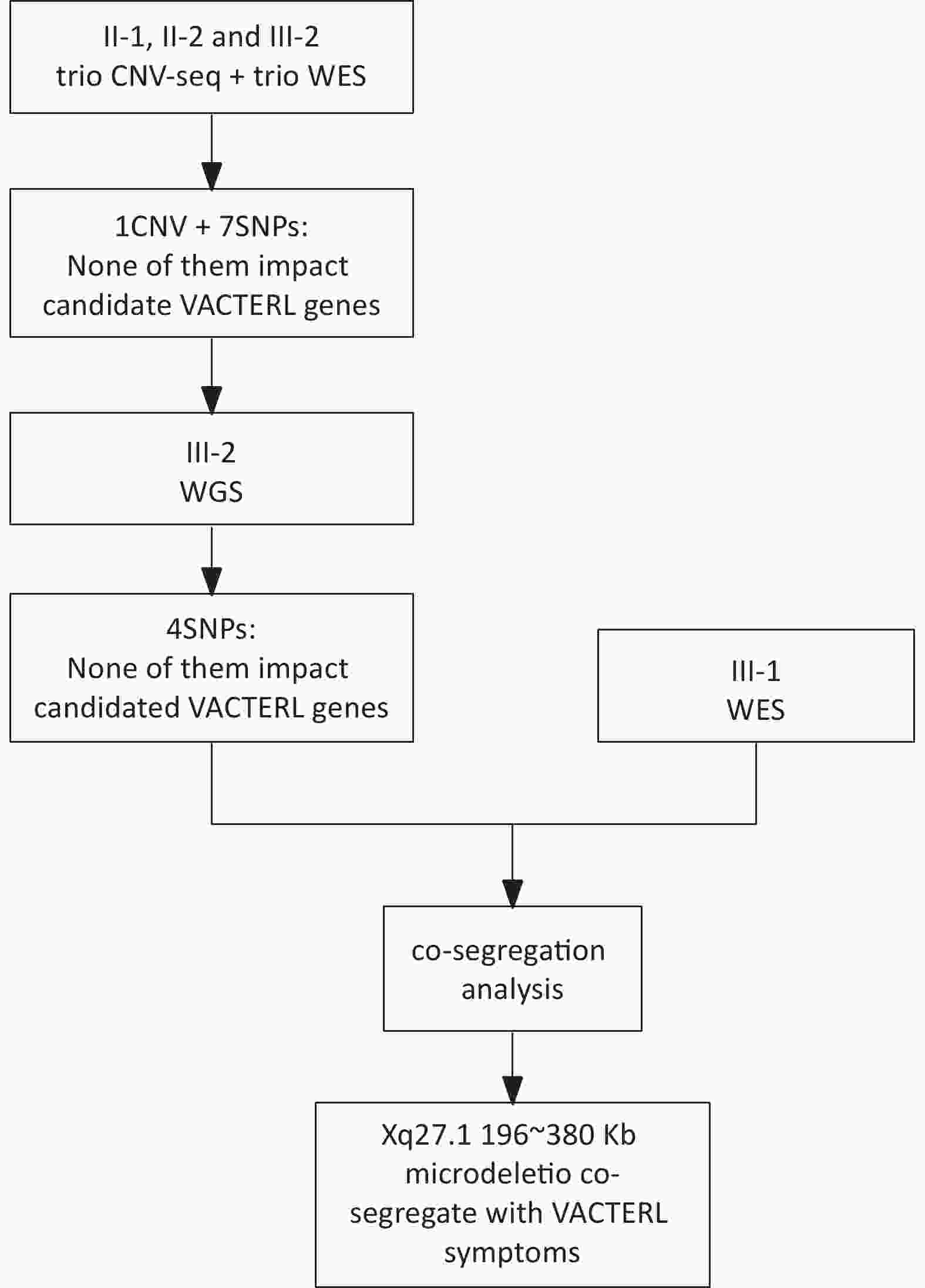
Figure 2. The flow chart of the investigation procedure. WGS, whole-genome sequencing; WGS, whole-exome-sequencing
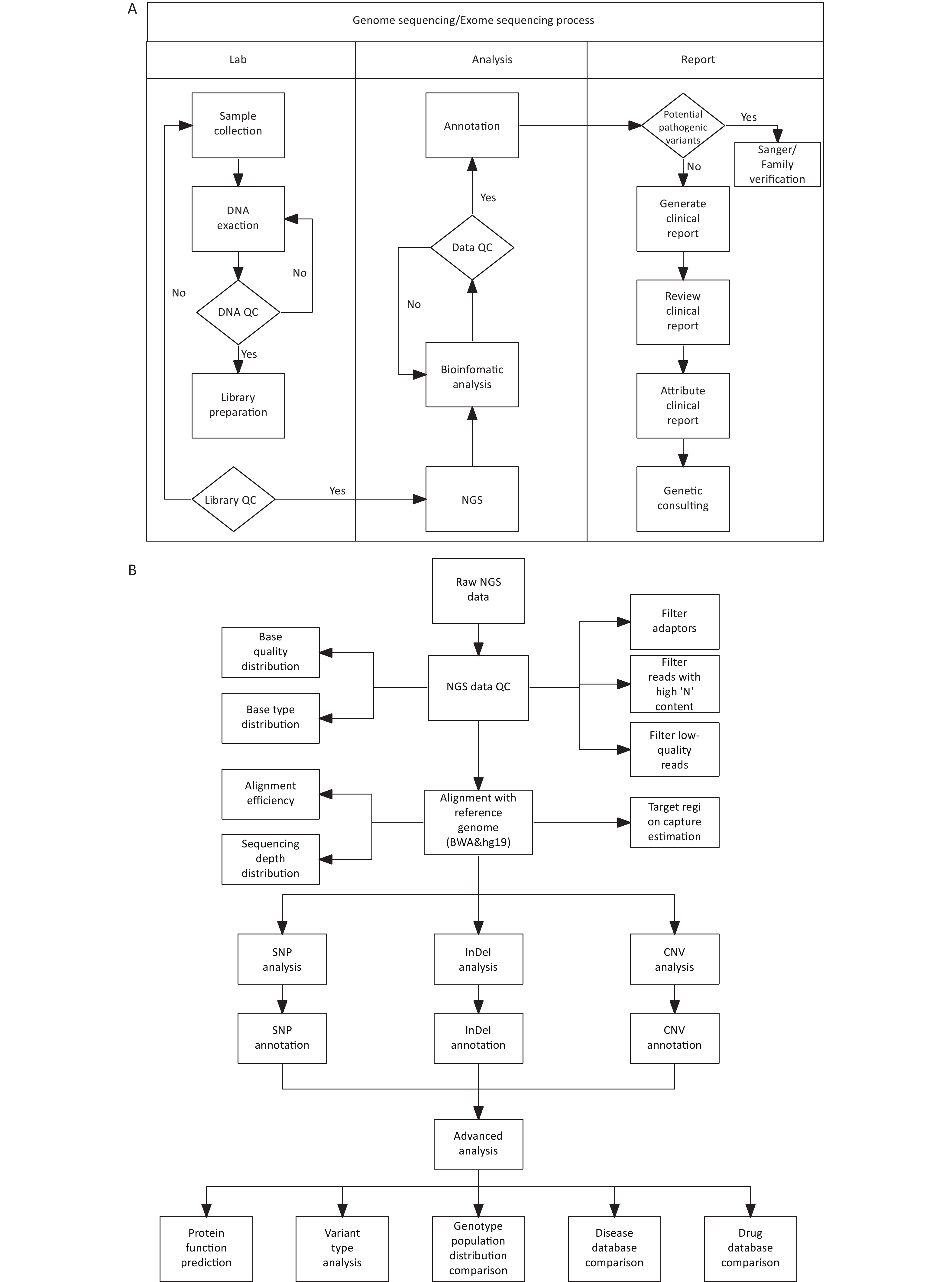
DNA library construction and sequencing were conducted at the Forevergen Biosciences Center (Guangzhou, China) using the MGI Easy Universal DNA Library Prep Set. Blood samples from unaffected individuals in the family were used as controls. Quality control of the sequencing data was performed using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapter sequences were removed from raw sequencing data using cutadapt[8]. The resulting clean reads were aligned to the human reference genome hg19 using BWA[9] and further processed using SAMtools[10]. Variants in SNPs and small insertions/deletions were detected by using GATK[11]. Copy number alteration (CNVs) analysis was performed using the read-depth method using an in-house program. ANNOVAR (http://annovar.openbioinformatics.org/en/latest/) was used to annotate all variations (Figure 3).
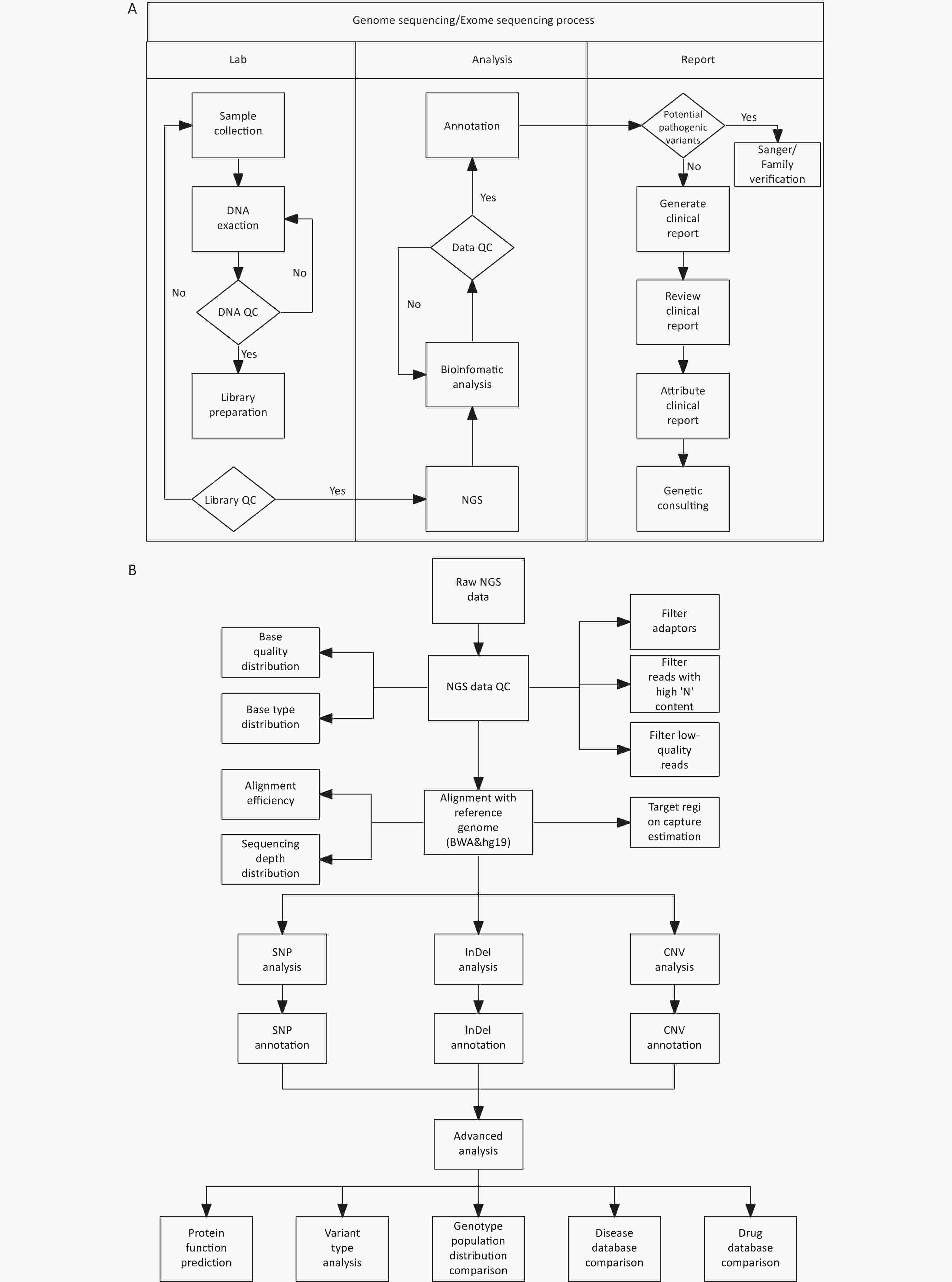
Figure 3. Procedures for genetic test and bioinformatics analysis.
All variations derived from sequencing, encompassing CNVs and SNPs, were interpreted in line with the ACMG guidelines, and each variation was determined to have uncertain clinical relevance. Following this, we conducted a co-segregation analysis of the family in question.
-
Seven family members were recruited in the study (Table 1), including three participants with the VACTERL association based on the family member’s description and medical records. All three affected individuals presented with unilateral renal absence, two with anal atresia (I-2 and III-2) and/or a single umbilical artery (II-2 and III-2), and one with an anovaginal fistula (II-2). All probands demonstrated normal height, posture, cardiovascular anatomy, and an intact esophagus and trachea, along with normal limb development. None of the patients presented vertebral, cardiac, or limb abnormalities. None of the participants had cognitive defects. None of the participants had any identified genetic etiology.
Table 1. Phenotypes of affected and unaffected members in the family
Family members Gender V A C TE R L Physical Abnormalities Cognitive Impairment I-1 - - - - - - - - - I-2 Female - + - - - - - - II-1 - - - - - - - - - II-2 Female - + - - + - - - III-1 - - - - - - - - - III-2 Male + + - - + - - - Note. V,vertebral anomalies; A, anal atresia; TE,tracheoesophageal malformations; R, renal dysplasia; L, limb abnormalities. Because I-2 passed away, we collected her medical characteristics based on her medical records and offspring’s descriptions (Table 2). I-2, with a left renal defect and anal atresia, delivered three children, including two daughters and one son. Her first daughter (II-2) was identified with a single umbilical artery and an absence of the left kidney during her fetal period. She presented with a congenital anovaginal fistula after birth and a congenital anovaginal fistula after birth. I-2 also gave birth to a healthy daughter (II-3) and a healthy son (II-4). Patient II-2 underwent fistula repair surgery and was cured in early childhood. II-2 also presented with the absence of a left fallopian tube and ovary. II-2 got married at 22 years of age and became pregnant four times after marriage. The first child of II-2 was a healthy girl (III-1). However, her second baby (III-2) was a son with a VACTERL association, including right renal agenesis and a single umbilical artery, reported by prenatal ultrasonography imaging and anal atresia reported after birth. The third and fourth children of II-2 were medically terminated in the second trimester because of oligohydramnios. However, none of these patients underwent genetic analysis. All family members, including the affected individuals, presented with a normal appearance and cognition.
Table 2. WGS and WES filtered CNV
Family number Gender Phenotype CHR Position Size Genotype I-1 Male − I-2 Female + II-1 Male − II-2 Female + X 140379030-140761877 −380 kb Heterzygote III-1 Female − III-2 Male + X 140590843-140786655 −196 kb Hemizygote Note. WES, whole-exome-sequencing; WGS,whole-genome sequencing; CNV, copy number-variation; CHR, chromosome -
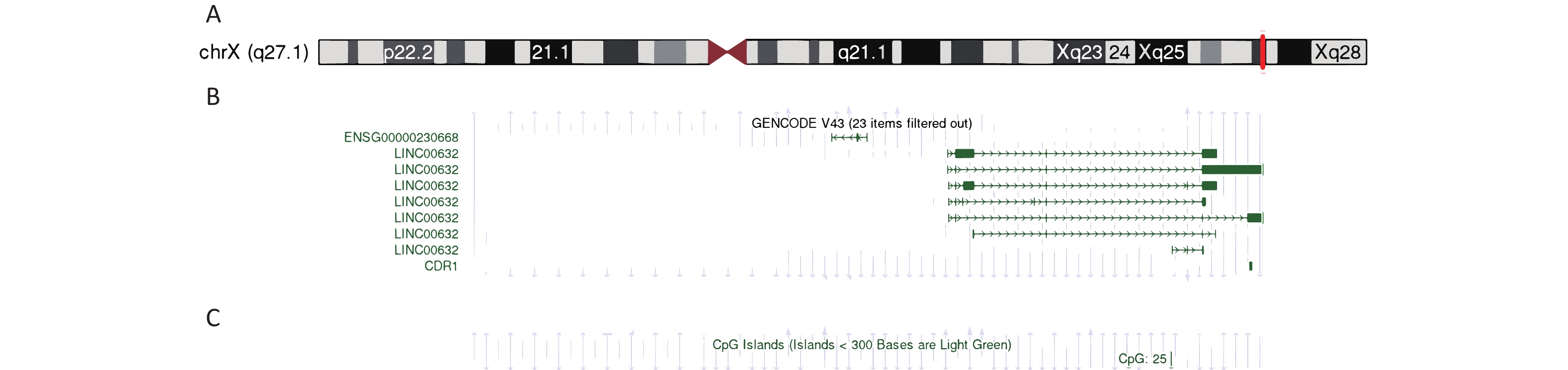
Four family members, including the living individuals affected by the disorder, underwent genetic analysis. The results of the whole-exome sequencing of the family and affected individuals are shown in Table 2. After processing and annotation of the WGS/WES/CNV-seq data, a microdeletion of approximately 196–380 kb on chromosome X co-segregating with disease symptoms was identified. More specifically, a -196 kb microdeletion in the Xq27.1 region (Figure 4A, 4B) was observed in individual III-2, whereas individual II-2 exhibited a -380 kb microdeletion at the same locus (Table 2).
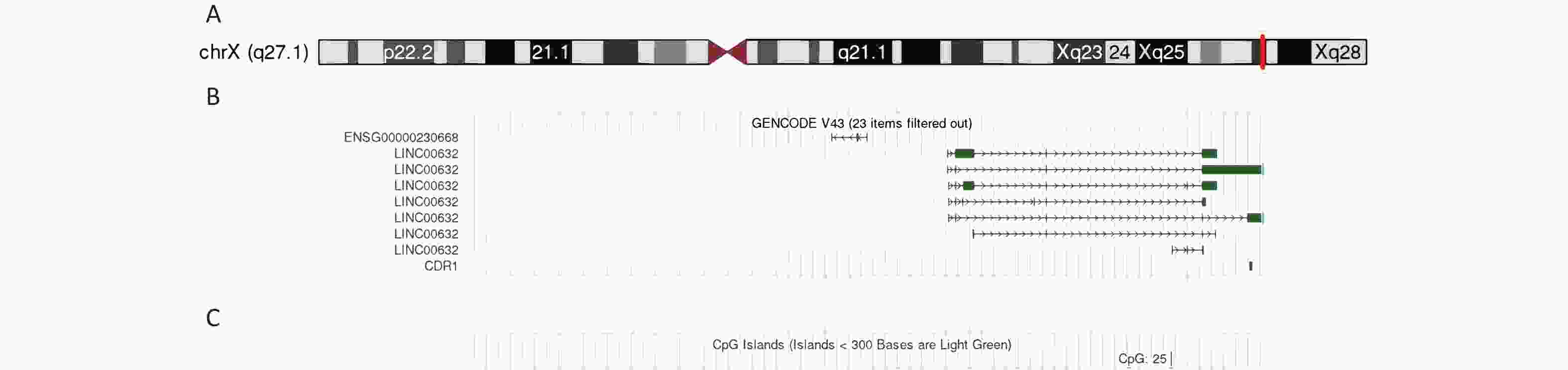
Figure 4. Genetic analysis of the Xq27.1 microdeletion.
The microdeletion region on Xq27.1, along with one CpG island, encompassed approximately 28% of the CDR1 sequence (Figure 4C).
Seven of eight family members, including four parents, two siblings, and five descendants of proband II-2, underwent co-segregation analysis. Clinical symptoms consistent with the VACTERL association were observed in three members. Notably, two of the three probands possessed identical Xq27.1 microdeletion. The family pedigree and associated parameters of the VACTERL association are shown in Figure 2. Our co-segregation analysis revealed a novel variant in the X chromosome present in two probands (individuals II-2 and III-2), a variant absent in unaffected family members.
-
In this study, we report a Chinese family manifesting VACTERL-like symptoms with a kidney-anal anomaly associated with a microdeletion in the Xq27.1 region. Our findings indicated an increased susceptibility to major component features among first-degree relatives of the proband. These results contribute to a deeper understanding of the genetic underpinnings of VACTERL without any overlap in clinical manifestations or genetic findings.
Although the VACTERL association is typically diagnosed when patients exhibit three or more component features (vertebral defect, anal atresia, cardiac malformation, tracheo-Esophageal fistula, renal anomalies, or limb defects). Patients with only two component features can be diagnosed as VACTERL-like[12]. Although both probands involved in our study presented with only two abnormal features qualifying for a VACTERL-like diagnosis[12], the characterization of congenital kidney anomalies and anal atresia shares a consistent inheritable genetic phenotype in this family. This finding raises concerns in both sporadic and familial reports on the association of VACTERLs with spinal malformations as the main clinical feature. The symptoms observed in this family may also indicate a localized developmental field defect[13] and appear to be an extended form of renal dysplasia[14,15]. In addition, a single umbilical artery was observed in both the probands’ fetuses, a unique feature that contributes to the complexity of their clinical profiles. Although a single umbilical artery is not typically considered a part of the VACTERL association or malformation status, our results suggest that a single umbilical artery indicates an increased risk of associated malformations and chromosomal abnormalities, leading to pregnancy complications[16]. Solomo et al.[3] also reported a relationship between a single umbilical artery and VACTERL, which is consistent with our results.
Recent studies have increasingly implicated genetic factors in the etiology of VACTERL, particularly in cases presenting with vertebral phenotypes. Certain monogenic diseases, including Fanconi anemia, complementation group A[6] and Alagille syndrome 1[17], exhibit at least two overlapping manifestations of the VACTERL association[18]. Trisomy 18 or 21 and CHARGE syndrome, which are often accompanied by fistula/esophageal atresia, have been reported as heritable components in individuals with VACTERL associations[19,20]. Several candidate gene variants have been associated with VACTERL[21]. Although the genetic underpinnings of the VACTERL association are yet to be fully elucidated, previous research has suggested that these candidate genes should be incorporated into diagnostic protocols and genetic studies about the VACTERL association[13]. Furthermore, the findings indicate that VACTERL syndrome should be considered a diagnosis of exclusion made only after a thorough clinical evaluation and careful consideration of genetic testing for syndromes with overlapping characteristics.
In the present study, we identified a microdeletion in the Xq27.1 region as a novel hereditary component of VACTERL association within our research cohort. While X-linked VACTERL associations have been sporadically reported in instances of VACTERL with Hydrocephalus Syndrome[6,22,23] and/or Fanconi anemia[22,24], the participants typically exhibited at least two of the three most common features (cardiac, renal, and upper limb malformations) and may also present with hematological and/or oncological anomalies. Interestingly, the probands in our study did not exhibit any of the aforementioned phenotypes except for renal malformations. We hypothesize that this discrepancy may be due to differing regions of involvement on the X chromosome (Xp22.3 and Xq12)[6,22]. Another region linked to the X-linked VACTERL association was the ZIC3 variant in Xq26.2, which is commonly associated with anal atresia[25]. These reported VACTERL presentations are consistent with the model reported by Toriello et al.[26], in which mitotic errors led to developmental defects mediated by the elimination of aneuploid clones, particularly midline defects, suggesting a possible cause of VACTERL.
To date, there have been no reports linking the Xq27.1 region with the VACTERL association, making this the second known instance of an X chromosome region being implicated in the VACTERL association. Several genes in this region play key roles in embryonic development and hypoparathyroidism[21,27]. Since there is no prior literature associating this Xq27.1 region with VACTERL syndrome, we propose that the deletion fragment in this region warrants further investigation.
The SOX3 gene, located near and upstream of the deleted region, is associated with congenital anomalies of the kidneys and urinary tract[28] but has not been mentioned in any VACTERL report. Furthermore, we did not find any genetic variants involved in the VACTER/VACTERL-like phenotypes. Although the implications of the Xq27.1 microdeletion remain uncertain, the deletion was the only genetic discrepancy observed between family members with and without VACTERL phenotypes, leading us to consider it as a candidate for VACTERL association based on co-segregation analysis.
In addition to SOX3, CDR1 is also involved in the Xq27.1 microdeletion. Variants or duplications of SOX3 have been linked to central congenital hypothyroidism[29] and prostatic utricle formation[28], but no studies have explored the association between SOX3 or CDR1 and VACTERL. The function of deleted methylation sites in this context remains poorly understood. Both probands exhibited the same methylation site deletion in human umbilical vein endothelial cells (HUVEC), which may account for the shared phenotype of a single umbilical artery. The roles of other deleted methylation sites remain unclear.
Given the variable phenotypes of the VACTERL association[30], the importance of DNA methylation, an essential epigenetic mechanism that regulates gene expression during embryonic development, should not be overlooked during prenatal diagnosis. Clinical genetic testing often results in variants of uncertain significance (VUS) that can be challenging for both clinicians and patients. Therefore, continuous collection, reporting, and exchange of gene testing results and clinical phenotype cases, rather than exclusively focusing on large databases and meta-analyses that may exclude rare symptoms for various reasons, can significantly improve the quality of clinical genetic testing databases[31].
Traditionally reliant on symptomatology since its inception in 1973, the approach to VACTERL diagnosis is now evolving towards integrating genetic diagnostics. This shift paves the way for more precise genetic studies and phenotypic subgrouping, particularly in cases exhibiting two or more malformations (VACTERL-like) or three or more malformations (typical VACTERL). Utilizing genetic algorithms such as CGH and NGS could unveil phenocopies and molecular anomalies, challenging the broad categorization inherent in VACTERL diagnosis and advocating for its refinement. This strategy emphasizes the importance of a nuanced diagnostic framework that may ultimately refine the use of the VACTERL acronym, allowing for more accurate patient classification and improving the diagnosis, prevention, and treatment of VACTERL in clinical practice.
-
We identified the first reported family in which four members with a kidney-anal anomaly fulfilled the diagnostic criteria for VACTERL-like association, with a ~196 kb microdeletion in the Xq27.1 region of the X chromosome. Our findings not only offer novel genetic insights that may improve the diagnosis of VACTERL but also indicate the potential involvement of this Xq27.1 microdeletion in the development of renal anomalies and anal dysplasia.
-
This study was approved by the Ethics Committee of Beijing Hospital. All the participants provided written informed consent. All work described in this study was conducted in accordance with the Declaration of Helsinki and the Code of Ethics of the World Medical Association. Only unidentified data were used in this study.
-
LI Min: Conceptualization, project administration, resources, supervision, visualization, writing the original draft, writing the review, and editing. ZHANG Yu Lan: Data curation, formal analysis, supervision, validation, and visualization. Zhang Kai Li: Investigation. LI Ping Ping: Investigation. LYU Yu Han: Investigation. LIANG Ya Xin: Investigation.
doi: 10.3967/bes2024.055
Microdeletion on Xq27.1 in a Chinese VACTERL-Like Family with Kidney and Anal Anomalies
-
Abstract:
Objective VATER/VACTERL-like association is associated with adverse pregnancy outcomes. Genetic evidence of this disorder is sporadic. In this study, we aimed to provide genetic insights to improve the diagnosis of VACTERL. Methods We have described a Chinese family in which four members were affected by renal defects or agenesis, anal atresia, and anovaginal fistula, which is consistent with the diagnosis of a VACTERL-like association. Pedigree and genetic analyses were conducted using genome and exome sequencing. Results Segregation analysis revealed the presence of a recessive X-linked microdeletion in two living affected individuals, harboring a 196–380 kb microdeletion on Xq27.1, which was identified by familial exome sequencing. Genome sequencing was performed on the affected male, confirming a -196 kb microdeletion in Xq27.1, which included a 28% loss of the CDR-1 gene. Four family members were included in the co-segregation analysis, and only VACTERL-like cases with microdeletions were reported in X27.1. Conclusion These results suggest that the 196–380 kb microdeletion in Xq27.1 could be a possible cause of the VATER/VACTERL-like association. However, further genetic and functional analyses are required to confirm or rule out genetic background as the definitive cause of the VACTERL association. -
Key words:
- Prenatal diagnosis /
- VACTERL /
- whole-genome sequencing /
- whole-exome sequencing /
- X-linked
注释:1) CONFLICT OF INTEREST: -
Figure 1. Family pedigree analysis.
Pedigree showing familial relationships of eight individuals. Squares denote men, circles denote women, and dots denote abortions or stillbirths (sex-unspecified). Black symbols indicate patients with VACTERL associations (I-2, II-2, III-2, and III-5 renal defects; I-2, III-2, and III-5 anal atresia; II-2 anovaginal fistula; and III-3, III-4, and III-5 oligohydramnios). Arrows indicate the samples included in the genetic analysis.
Figure 2. The flow chart of the investigation procedure. WGS, whole-genome sequencing; WGS, whole-exome-sequencing
Figure 3. Procedures for genetic test and bioinformatics analysis.
(A) WGS/WES process chart (including CNV-seq as low-depth WGS); (B) Bioinformatic analysis procedure. NGS, next-generation sequencing; QC, quality control; WGS, whole-genome sequencing;WGS, whole-exome-sequencing; CNV, copy number-variation
Figure 4. Genetic analysis of the Xq27.1 microdeletion.
(A) The position of Xq27.1 microdeletion; (B) The content of missing region; (C) The position of CpG island.
Table 1. Phenotypes of affected and unaffected members in the family
Family members Gender V A C TE R L Physical Abnormalities Cognitive Impairment I-1 - - - - - - - - - I-2 Female - + - - - - - - II-1 - - - - - - - - - II-2 Female - + - - + - - - III-1 - - - - - - - - - III-2 Male + + - - + - - - Note. V,vertebral anomalies; A, anal atresia; TE,tracheoesophageal malformations; R, renal dysplasia; L, limb abnormalities.  下载: 导出CSV
下载: 导出CSV
Table 2. WGS and WES filtered CNV
Family number Gender Phenotype CHR Position Size Genotype I-1 Male − I-2 Female + II-1 Male − II-2 Female + X 140379030-140761877 −380 kb Heterzygote III-1 Female − III-2 Male + X 140590843-140786655 −196 kb Hemizygote Note. WES, whole-exome-sequencing; WGS,whole-genome sequencing; CNV, copy number-variation; CHR, chromosome
下载: 导出CSV
-
[1] Solomon BD. The etiology of VACTERL association: current knowledge and hypotheses. Am J Med Genet C Semin Med Genet, 2018; 178, 440−6. doi: 10.1002/ajmg.c.31664 [2] Carli D, Garagnani L, Lando M, et al. VACTERL (vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, cardiac defects, renal and limb anomalies) association: disease spectrum in 25 patients ascertained for their upper limb involvement. J Pediatr, 2014; 164, 458-62. e2. [3] Solomon BD. VACTERL/VATER association. Orphanet J Rare Dis, 2011; 6, 56. doi: 10.1186/1750-1172-6-56 [4] Amelot A, Cretolle C, de Saint Denis T, et al. Spinal dysraphism as a new entity in V. A. C. TE. R. L syndrome, resulting in a novel acronym V. A. C. TE. R. L. S. Eur J Pediatr, 2020; 179, 1121−9. doi: 10.1007/s00431-020-03609-4 [5] van de Putte R, van Rooij IALM, Marcelis CLM, et al. Spectrum of congenital anomalies among VACTERL cases: a EUROCAT population-based study. Pediatr Res, 2020; 87, 541−9. doi: 10.1038/s41390-019-0561-y [6] McCauley J, Masand N, McGowan R, et al. X-linked VACTERL with hydrocephalus syndrome: further delineation of the phenotype caused by FANCB mutations. Am J Med Genet A, 2011; 155A, 2370−80. [7] Solomon BD, Pineda-Alvarez DE, Raam MS, et al. Evidence for inheritance in patients with VACTERL association. Hum Genet, 2010; 127, 731−3. doi: 10.1007/s00439-010-0814-7 [8] Kechin A, Boyarskikh U, Kel A, et al. cutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J Comput Biol, 2017; 24, 1138−43. doi: 10.1089/cmb.2017.0096 [9] Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009; 25, 1754−60. doi: 10.1093/bioinformatics/btp324 [10] Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics, 2009; 25, 2078−9. doi: 10.1093/bioinformatics/btp352 [11] DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet, 2011; 43, 491−8. doi: 10.1038/ng.806 [12] Lubinsky M. The VACTERL association: mosaic mitotic aneuploidy as a cause and a model. J Assist Reprod Genet, 2019; 36, 1549−54. doi: 10.1007/s10815-019-01485-y [13] Opitz JM. The developmental field concept in clinical genetics. J Pediatr, 1982; 101, 805−9. doi: 10.1016/S0022-3476(82)80337-5 [14] Buchta RM, Viseskul C, Sarto GE, et al. Familial bilateral renal agenesis and hereditary renal adysplasia. Z Kinderheilkd, 1973; 115, 111−29. doi: 10.1007/BF00440537 [15] McPherson E, Carey J, Kramer A, et al. Dominantly inherited renal adysplasia. Am J Med Genet, 1987; 26, 863−72. doi: 10.1002/ajmg.1320260413 [16] Murphy-Kaulbeck L, Dodds L, Joseph KS, et al. Single umbilical artery risk factors and pregnancy outcomes. Obstet Gynecol, 2010; 116, 843−50. doi: 10.1097/AOG.0b013e3181f0bc08 [17] Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet, 2012; 20: 251-7. [18] Chen Y, Liu Z, Chen J, et al. The genetic landscape and clinical implications of vertebral anomalies in VACTERL association. J Med Genet, 2016; 53: 431-7. [19] Brown AK, Roddam AW, Spitz L, et al. Oesophageal atresia, related malformations, and medical problems: a family study. Am J Med Genet, 1999; 85: 31-7. [20] van Lennep M, Singendonk MM, Dall’Oglio L, et al. Oesophageal atresia. Nature Reviews disease primers, 2019; 5: 26. [21] Winberg J, Gustavsson P, Papadogiannakis N, et al. Mutation screening and array comparative genomic hybridization using a 180K oligonucleotide array in VACTERL association. PLoS One, 2014; 9: e85313. [22] Mikat B, Roll C, Schindler D, et al. X-linked recessive VACTERL-H due to a mutation in FANCB in a preterm boy. Clinical Dysmorphology, 2016; 25: 73-6. [23] Herman TE, Siegel MJ. Vacterl-H syndrome. Journal of perinatology, 2002; 22: 496-8. [24] Fiesco-Roa MO, Giri N, McReynolds LJ, et al. Genotype-phenotype associations in Fanconi anemia: a literature review. Blood reviews, 2019; 37, 100589. doi: 10.1016/j.blre.2019.100589 [25] Hilger AC, Halbritter J, Pennimpede T, et al. Targeted resequencing of 29 candidate genes and mouse expression studies implicate ZIC3 and FOXF1 in human VATER/VACTERL association. Hum Mutat, 2015; 36: 1150-4. [26] Toriello HV, Higgins JV. X-linked midline defects. Am J Med Genet, 1985; 21: 143-6. [27] Bowl MR, Nesbit MA, Harding B, et al. An interstitial deletion-insertion involving chromosomes 2p25. 3 and Xq27. 1, near SOX3, causes X-linked recessive hypoparathyroidism. J Clin Invest, 2005; 115: 2822-31. [28] Tasic V, Mitrotti A, Riepe F, et al. Duplication of the gene in an sry-negative 46, XX male with associated congenital anomalies of kidneys and the urinary tract: Case report and review of the literature. Balkan Journal of Medical Genetics, 2019; 22: 81-8. [29] Wei J, Liu C, Zhang M, et al. Duplication of SOX3 in an SRY-negative 46, XX male with prostatic utricle: case report and literature review. BMC Med Genomics, 2022; 15: 188. [30] Chen CP, Shih JC, Chang JH, et al. Prenatal diagnosis of right pulmonary agenesis associated with VACTERL sequence. Prenatal Diagnosis: Published in Affiliation With the International Society for Prenatal Diagnosis, 2003; 23: 515-8. [31] Amendola LM, Dorschner MO, Robertson PD, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res, 2015; 25: 305-15. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 610
- HTML全文浏览量: 236
- PDF下载量: 40
- 被引次数: 0

 Quick Links
Quick Links